| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://cr.elmerpub.com |
Review
Volume 16, Number 2, April 2025, pages 86-101

Advances in Pathophysiological Mechanisms of Degenerative Aortic Valve Disease
Ya Lu Yua, Qin Jianga, b, c, d
aSchool of Medicine, University of Electronic Science and Technology of China, 610072 Chengdu, Sichuan, China
bDepartment of Cardiac Surgery, Sichuan Provincial People’s Hospital, Affiliated Hospital of University of Electronic Science and Technology, 610072 Chengdu, Sichuan, China
cUltrasound in Cardiac Electrophysiology and Biomechanics Key Laboratory of Sichuan Province, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, 610072 Chengdu, Sichuan, China
dCorresponding Author: Qin Jiang, Department of Cardiac Surgery, Sichuan Provincial People’s Hospital, Affiliated Hospital of University of Electronic Science and Technology, 610072 Chengdu, Sichuan, China
Manuscript submitted November 22, 2024, accepted February 6, 2025, published online February 18, 2025
Short title: Advances in Mechanisms of DAVD
doi: https://doi.org/10.14740/cr2012
| Abstract | ▴Top |
Degenerative aortic valve disease (DAVD) represents the most prevalent valvular ailment among the elderly population, which significantly impacts their physical well-being and potentially poses a lethal risk. Currently, the underlying mechanisms of DAVD remain incompletely understood. While the progression of this disease has traditionally been attributed to degenerative processes associated with aging, numerous recent studies have revealed that heart valve calcification may represent a response of valve tissue to a specific initiating factor, involving the interaction of various genes and signaling pathways. This calcification process is further influenced by a range of factors, including genetic predispositions, environmental exposures, metabolic factors, and hemodynamic considerations. Based on the identification of its biomarkers, potential innovative therapeutic targets are proposed for the treatment of this complex condition. The present article primarily delves into the underlying pathophysiological mechanisms and advancements in diagnostic and therapeutic modalities pertaining to this malady.
Keywords: Degenerative heart valve disease; Pathophysiological mechanisms; Diagnostic; Treatment; Prognosis
| Introduction | ▴Top |
Degenerative aortic valve disease (DAVD) is a valvular heart disease prevalent in the elderly population that has superseded rheumatic valve disease as the principal clinical manifestation of heart valve disease [1]. The dysfunction of the aortic valve typically arises from degeneration, fibrosis, and calcification, and the progression of this condition ultimately culminates in aortic valve stenosis and incomplete closure [2], which is a significant factor contributing to decreased activity tolerance, heart failure (HF), arrhythmia, recurrent hospitalizations, and mortality among the elderly population [3]. The frequently encountered form of DAVD is calcific aortic valve disease (CAVD) in clinical practice, which is a progressive dystrophic calcification of the aortic valve that eventually advances to aortic stenosis (AS) [4]. In the past, this procedure was perceived as a degenerative and senile-like mechanism due to the time-dependent attrition of the leaflets, leading to passive calcium deposition [5]. But immunohistological studies dating back to 30 years ago had demonstrated that the pathological mechanisms underlying aortic valve degeneration are similar to those active in the process of atherogenesis [6]. These similarities include the disruption of valve endothelial architecture, the localized infiltration of inflammatory cells, such as monocytes and T lymphocytes, the secretion of inflammatory cytokines, the proliferative activity of valve interstitial cells, the restructuring of the extracellular matrix, and the calcification process [7, 8]. Up until now, the precise pathogenesis of DAVD has remained incompletely understood. It is known to be associated with genetic factors, environmental conditions, metabolic processes, and hemodynamic mechanisms. This review aims to concentrate on the investigation of pathophysiological mechanisms, ranging from the molecular basis to the clinical significance, and to provide comprehensive insights into the current status of the disease.
| Epidemiology | ▴Top |
In recent years, with the aging population, the incidence and related health burden of DAVD are increasing. From 1990 to 2017, across 195 countries and territories, the number of disability-adjusted life years (DALYs) attributable to cardiovascular, atherosclerotic, and related diseases (CAVD) increased by 101%. Moreover, the age-standardized rate of DALYs due to CAVD is observed to be highest in high-income nations [9]. In the 2021 global epidemiological survey on valvular heart disease, DAVD emerged as a prevailing worldwide concern, exhibiting notable regional disparities. Its impact remains substantial even in affluent regions like Oceania, Europe, and North America [10]. A recent survey investigating the prevalence and causes of valvular heart disease in China examined 31,499 individuals, among whom 1,309 were diagnosed with cardiac valvular disease. Of these cases, 21.3% were attributed to degenerative causes, with the incidence rising as the age of the subjects increased [11].
In the population over 80 years of age, this figure is expected to approximately double, reaching 8-10% [12]. Recent years’ data indicate that up to 13% of individuals aged 65 and older are affected by DAVD [13]. According to retrospective and prospective studies, the key risk factors for developing CAVD also include male gender (women have an increased risk after postmenopausal period) [14], hypertension (primarily manifesting as elevated systolic blood pressure and pulse pressure) [15], hyperlipidemia (particularly elevated levels of plasma lipoprotein(a) (Lp(a)) and low-density lipoprotein (LDL) cholesterol) [16], diabetes and genetic history [17].
DAVD may result in a range of severe complications, including HF, arrhythmia (particularly atrial fibrillation) and valvular endocarditis, among others [18]. A previous study showed that the 5-year survival rate was less than 50% [19]. In particular, the trends in DAVD-related HF from 1990 to 2019 and the forecast to 2030 in 20 countries, indicate that the prevention and treatment of DAVD-related HF remains an urgent and unaddressed global public health challenge. Variety countries must develop comprehensive health policies that prioritize early screening, early prevention, and early treatment to effectively address the significant burden of DAVD-related HF [20].
| Valvular anatomy | ▴Top |
The aortic valve is composed of three semilunar leaflets: the left, right, and non-coronary. These leaflets are anchored to the aortic annulus and open during the heart’s contraction phase to permit blood flow from the left ventricle into the aorta. Conversely, they close when the heart relaxes to prevent the backflow of blood [21]. Degenerative alterations can cause the valve to stiffen and become calcified, resulting in valve dysfunction. At the cellular level, the normal cardiac valve consists of the valvular interstitial cells (VICs), valvular endothelial cells (VECs) and extracellular matrix (ECM) [22]. In healthy valves, ECM is meticulously arranged into three distinct layers according to blood flow, including the fibrosa, ventricularis and spongiosa [23]. The fibrous membrane layer, situated proximally to the effluent surface, comprises densely packed type I and III collagen fibers, accountable for maintaining the mechanical integrity [24]. Conversely, the ventricular layer, positioned on the opposing inflow surface, is abundant in elastin, effectively distributing the hydrodynamic pressure and ensuring flexibility and resilience within the cardiac cycle [25]. Interposed between these two layers is the cavernosal layer, which exhibits a loosely organized central connective tissue enriched with proteoglycans (PGs) and glycosaminoglycans (GAGs) [26]. This arrangement facilitates the rearrangement of collagen and elastic layers during the cardiac cycle, accommodating the relative movement of adjacent layers [5]. The VECs cover the surface of the heart valve in direct contact with the blood, serving as a physical barrier between the VICs and the hemodynamic environment, and have been shown to influence the behavior of the VIC, modulating its phenotype [27, 28]. VICs are distributed sporadically within the valve interior, serving as the essential cellular constituents of the valve leaflet [29]. They are also capable of secreting a diverse array of ECM components, such as collagen and elastic fibers, thereby ensuring that the free collagen content within the valve remains at a relatively low level [30]. VICs possess the capability to transform into numerous phenotypic expressions, facilitating their adaptability to varying physiological environments [31, 32]. The valve structure (Fig. 1) illustrates that DAVD originates from ECM remodeling, which is induced by VICs and VECs under pathological conditions.
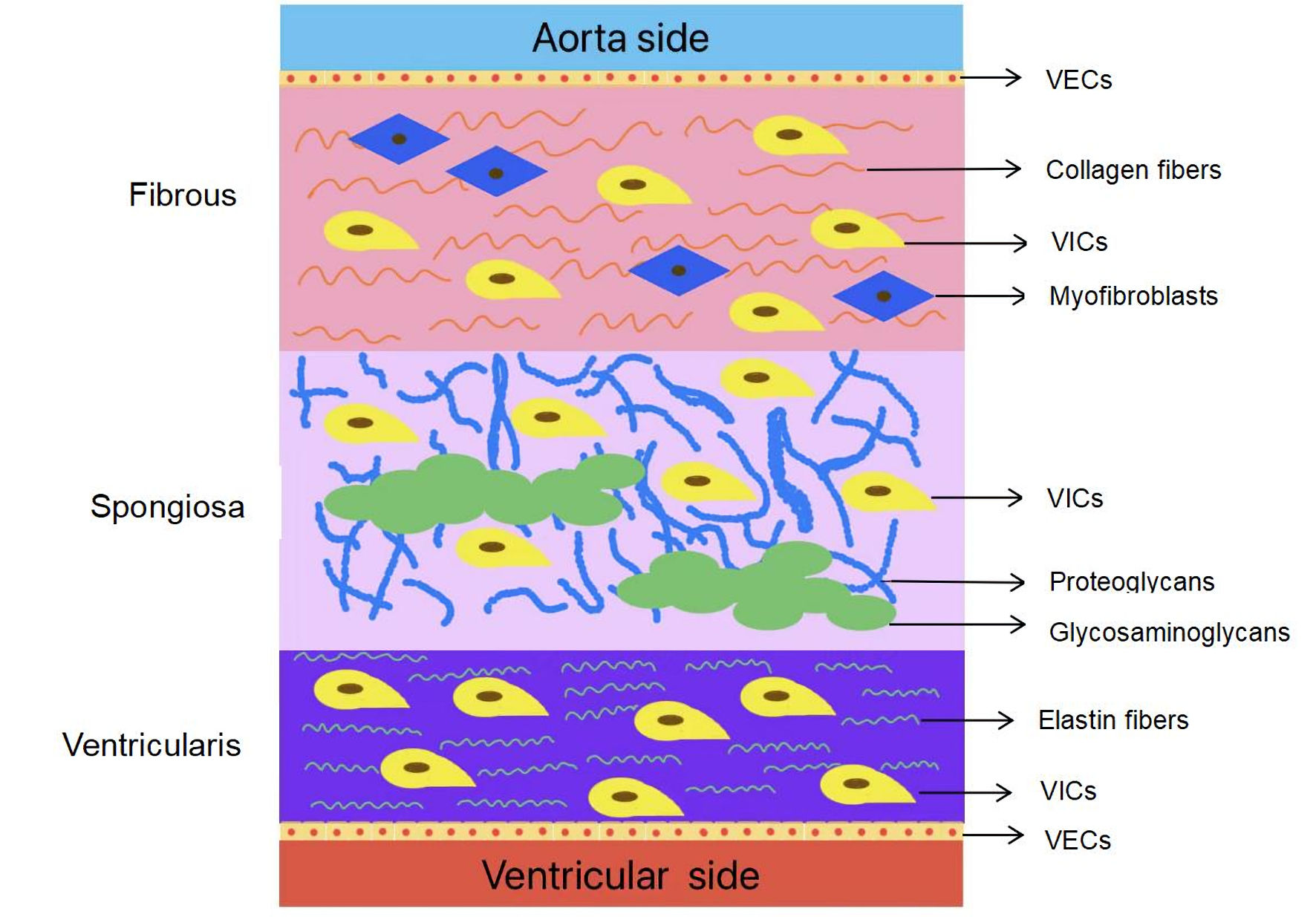 Click for large image | Figure 1. Anatomy of the normal human aortic valve. VICs: valvular interstitial cells; VECs: valvular endothelial cells. |
| Pathophysiological Mechanisms | ▴Top |
Cellular and molecular mechanisms
The pathophysiology of DAVD encompasses a range of cellular and molecular alterations, which are evident in the degeneration of valve structure, calcification, and cellular apoptosis.
Degeneration and apoptosis of the heart valve cells
From Figure 1, which shows the main components of the aortic valve, VICs have the ability to maintain the structural integrity and function of the valve under normal physiological conditions. However, in the event of alterations in valve homeostasis, including lipid oxidative stress, mechanical stress, and inflammatory cytokines, cardiomyocytes initiate the activation of VICs, ultimately resulting in their transformation into activated VICs [33, 34]. In its normal state, upon completion of the repair function for damaged valve tissue, activated VICs transition to the static phase or undergo spontaneous apoptosis to preserve the homeostasis of the valve tissue [35]. However, in the event that the activator remains persistent, VICs will continue to undergo differentiation into myofibroblasts and osteoblast-like cells, which are primarily accountable for the progression of valvular fibrosis and calcification, respectively [36].
Studies have indicated that VICs can be stimulated by inflammatory mediators, leading to diminished proliferative capabilities in fibroblasts. Concurrently, it has been found that an elevated propensity for apoptosis accelerates the degenerative processes within the valve [37]. Prior studies have established that an imbalance in the expression of some inflammatory mediators, such as tumor necrosis factor-α (TNF-α) and interleukin (IL) family, is potentially contributory to the development of DAVD [38]. TNF-α is an inflammatory factor secreted by numerous types of cells, including macrophages and T cells, and the existence of oxidized Lps within the valve promotes activated macrophages to express an increased amount of TNF-α [39]. TNF-α can activate signaling pathways such as cAMP/PKA to initiate calcification and promote valve fibrosis [40]. The potential involvement of the MAPK/ERK cascade and TNF-α interaction in the differentiation of the osteogenic phenotype in VICs is also noteworthy [41]. There are two cytokines in the IL family: one pro-inflammatory and one anti-inflammatory. IL-17 induces valvular endothelial inflammation and aggravates CAVD, while intracellular IL-37 exerts an anti-inflammatory effect through a nucleus-targeting mechanism [42, 43]. Recently, Candellier et al showed that indoxyl-sulfate activation of the AhR-NF-κB pathway promotes IL-6 secretion and the subsequent osteogenic differentiation of human VICs from the aortic valve [44]. IL-18, via the NF-κB pathway, is capable of facilitating VICs differentiation and triggering the production of TNF-α. This process creates a positive feedback loop, leading to the sustained generation of inflammatory cytokines and the exacerbation of osteogenic alterations. Ultimately, these effects contribute to the advancement of DAVD progression [45]. In this case, proapoptotic factors (e.g., cytochrome C and Bax) are upregulated, while the expression of anti-apoptotic factors (e.g., Bcl-2) decreases, leading to the activation of the apoptotic program and ultimately accelerating the degenerative changes of the valve [46].
Inflammatory factors not only stimulate apoptosis but also facilitate the breakdown of collagen, disrupting the maintenance of the ECM. This mechanism could result in structural alterations of the valve, potentially culminating in valvular dysfunction. The extensive remodeling of the ECM is accompanied by a series of cellular modifications throughout the process of DAVD [47]. This remodeling is characterized by the increased activity of various matrix metalloproteinases (MMPs), such as MMP-1 (fibroblast type collagenase), MMP-3(stromelysin-1), MMP-9 (gelatinase B), and MMP-13 (collagenase-3) [48], and the production and dysregulation of cathepsins and other proteolytic enzymes, as well as alterations in collagen [49]. Yu et al had showed that thrombospondin 2 expression was upregulated in CAVD and positively correlated with ECM-related (MMP-2 and MMP-13) factors in the process of osteogenic differentiation [50]. As MMPs promote valve remodeling and calcium in CAVD, studies with more potent and specific inhibitors are needed to establish any potential role of MMP inhibition in CAVD treatment [51].
The biological mechanism of the valve calcification
Valve calcification represents a crucial characteristic of DAVD, involving intricate biological processes primarily centered on the transformation of valvular cells. The osteogenic potential of endothelial-mesenchymal transition (EndMT) has been proposed as a mechanism underlying DAVD, primarily through the interaction and transformation of VECs and VICs [52]. EndMT plays an important pathophysiological role in the development of the embryonic cardiovascular system, cardiac fibrosis, atherosclerosis, and pulmonary hypertension [53-55]. The role of EndMT in adult valves remains enigmatic, yet it is believed to be a reaction to mechanical and biochemical stimuli that result in the remodeling of the underlying tissue [26]. Certain transcriptional factors (TFs) that have been implicated in the pivotal role of EndMT during valvular development may likewise become activated in the context of DAVD, potentially exerting pathological functions through the mediation of EndoMT [56]. In recent study, dual-specificity phosphatase 1 (DUSP1) potentially exerts a safeguarding function against the valve in inflammatory responses and mechanical stress, and pyruvate dehydrogenase kinase-4 (PDK-4) holds a pivotal position in regulating metabolic processes, EndMT and osteogenic transformation, presenting novel targets for future interventional strategies focused on EndMT.
During the development of DAVD, valve cells display remarkable characteristics of a bone-like transformation. VICs trigger the expression of genes associated with bone formation within a pathogenic setting, such as osteocalcin and bone sialoprotein (BSP) [57]. Osteocalcin serves as a distinctive indicator of cellular osteogenic transformation, playing a crucial role in bone formation and calcium metabolism [58]. The expression of BSP increases during the process of calcification, facilitating the deposition of calcium salts and augmenting calcium accumulation within the valve [59]. Proinflammatory factors play an important role in the regulation of the bone-like transition of VICs, especially the transforming growth factor β (TGF-β) and certain fatty acids. The TGF-β signaling pathway induces bone-like phenotypic alterations in VICs, triggering the expression of genes like osteocalcin and hastening the development of calcified nodules [60]. The synergistic effect of sphingosine 1-phosphate and lipopolysaccharide (LPS) signaling enhances angiogenesis and osteogenic responses in VICs [61]. Certain saturated fatty acids can induce the production of proinflammatory factors by VICs, such as TNF-α and IL-6, promoting the development of valve calcification through bone morphogenetic protein (BMP), Notch, Wnt/β-catenin, and NF-κB [39, 62]. Calcium deposition and alterations in membrane structure can lead not only to suboptimal valve coupling but may also result in valve stenosis and degeneration. These conditions can ultimately impair the heart’s pumping function and systemic blood flow dynamics.
Genes and the signaling pathways
The core feature of DAVD disease is the calcification of the valves, this process involves the complex regulation of multiple genes and signaling pathways. Several important genes and signaling pathways are proposed here.
Changes in expression of the associated genes
In recent years, more and more studies have shown that microRNA (miRNA) and long non-coding RNA (lncRNA) in non-coding RNA are closely related to DAVD, via promoting osteogenesis and calcification by targeting many genes [63, 64]. For example, Zheng et al [65] successfully identified the differentially expressed circular RNAs (circRNAs), lncRNAs, miRNAs, and mRNAs in CAVD, and subsequently established a comprehensive circRNA/lncRNA-miRNA-mRNA interaction network. Following rigorous verification using independent data sets and quantitative reverse transcription polymerase chain reaction (qRT-PCR) techniques, a final network encompassing hsa-circ-0073813/hsa-circ-0027587, hsa-miR-525-5p, and SPP1/HMOX1/CD28 was firmly established [65].
MiRNAs constitute a distinct class of short non-coding RNAs, approximately 22 nucleotides in length, which exert crucial functions in the post-transcriptional regulation of gene expression, involving in the occurrence and development of various cell biological processes, such as apoptosis, angiogenesis, inflammation, and ischemic preconditioning [66-69]. Genetic testing found that miRNA was differentially expressed in normal and calcified valves, which could promote or inhibit the differentiation of VICs to osteogenic-like phenotype and regulate the progression of DAVD [70]. It has been demonstrated that in DAVD, myocardial fibrosis (MF) is associated with regional and global strain alterations. Plasmatic miRNA-21 has been found to be directly related to MF and associated with left atrium (LA) structural and functional impairment. And miRNA-21 is a biomarker associated with MF in pressure overload. Study had found a comprehensive algorithm for detection of MF using a combinatorial approach of biological and functional markers [71]. The miR-125b is a potential regulator of the chemokine CCL4 in macrophages, and both exhibit upregulation in DAVD [72]. The direct target of miR-138 is FOXC1, and it has been observed that the overexpression of FOXC1 facilitates the osteogenic differentiation of VICs. Consequently, miR-138 impedes aortic valve calcification (AVC) by suppressing the osteogenic differentiation of VICs, indicating a potential focal point for targeted therapeutic intervention [73]. Low expression levels of miR-214 can eliminate the transcription factors Sp7 and ATF4, which inhibit osteogenesis, leading to increased expression of downstream genes such as CHOP and BCL2L1, and promoting the development of DAVD [74]. miR-222 not only plays a role in regulating calcification but is also independently associated with atrial fibrillation (AF) in patients with DVHD [75]. The activation of miR-486 within the serine/threonine protein kinase signaling pathway leads to an upregulation of α-smooth muscle actin (α-SMA) expression and concurrent downregulation of Smurf2 expression, resulting in the downregulation of miR-204 levels, ultimately facilitating accelerated valve calcification [76]. The miR-195 and miR-582 exert inhibitory effects on the expression of endothelial nitric oxide synthase (eNOS), thereby regulating the release of nitric oxide (NO) through post-transcriptional targeting of nitric oxide synthase 3 (NOS3) [77]. Consequently, this regulatory mechanism suppresses the calcification process mediated by VICs, offering insights into the intricate regulatory mechanisms that govern endothelial function and vascular homeostasis [78].
LncRNA also plays a significant role in the calcification process. LncRNA is a non-coding RNA of more than 200 nucleotide units in length, involved in vascular aging and calcification by interacting with miRNA or directly binding to proteins, and may have a regulatory role in the pathogenesis of DAVD [79]. Based on target gene prediction and co-expression network construction, 12 lncRNAs (CDKN2B-AS1, AC244453.2, APCDD1L-DT, SLC12A5-AS1, TGFB3, AC243829.4, MIR4435-2HG, FAM225A, BHLHE40-AS1, LINC01614, AL356417.2, LINC01150) were identified as the hub cis- or trans-regulatory genes in the pathogenesis of DAVD [80]. LncRNA MALAT1 exhibits regulatory effects on the proliferation and calcification of VICs, exacerbating calcified aortic valve in conjunction with miR-195 [81]. The findings indicate that the lncRNA AFAP1-AS1 is crucial for facilitating the M1 polarization of macrophages and the osteogenic differentiation of VICs through the modulation of the miR-155/SMAD5 axis [81, 82]. The lncRNA OIP5-AS1 exerts its regulatory function by upregulating the miR-137 target gene TWIST1, alleviating the osteogenic differentiation of VICs and shedding light on the therapy for DAVD [83].
The Runt-related transcription factor (Runx2) is a pivotal regulator of osteogenesis and calcification. Elevated expression of Runx2 is linked to the bone-like transformation of VICs. In conjunction with the involvement of miRNAs and lncRNAs, it establishes the foundation for the development of calcification. Overexpression of Runx2 may cause osteogenic differentiation in DAVD patients on inhibiting miR-138 by regulating the effect of Wnt/β-catenin signaling [73]. The regulation of miR-30b on the expression of Runx2, Smad1, and caspase-3 has elucidated its function in modulating the calcification process of human aortic VICs under in vitro conditions [84]. LncRNA MALAT1 attenuated the inhibitory effect of Runx 2 and Smad 4, and enhanced the calcification-inducing factor TGF-β1 by sponging miR-204, leading to the accelerated transformation of VICs to osteogenic phenotype [82]. LncRNA H19 upregulates p38 and p65, which are pivotal components of the MAPK and NF-κB signaling pathways, respectively, enhances the expression of Runx 2 and BMP-2 within VICs, and suppresses the transcription of Notch1, facilitating the osteogenic differentiation of VICs [85].
The role of the important signaling pathways
Notch1 serves as a crucial protein within the Notch signaling pathway, playing a pivotal role in diverse biological processes of cardiomyocyte, such as cell proliferation, differentiation, and apoptosis [86]. In DAVD, Notch signaling modulates the behavior of VICs, encompassing proliferation, migration, and osteogenic differentiation [87]. In a past study, intercellular adhesion molecule-1 (ICAM-1) and its ligand LFA-1 stimulated the activation of Notch1, leading to the upregulation of BMP-2/4 expression. This process triggered the activation of the nuclear transcription factor NF-κB and the MAPK/ERK (1/2) cascade, ultimately resulting in enhanced alkaline phosphatase activity and the formation of calcification nodules [88]. Bacterial LPS or peptidoglycan (PG) triggers the engagement of Notch1 protein with its cognate ligand, resulting in the formation of NIC and the activation of numerous cellular signaling cascades pertinent to inflammation and calcification, including NF-κB, ERK 1/2, and JAK-STAT [89, 90].
TGF-β signaling pathway plays a pivotal role in tissue remodeling and healing, and it also exerts a substantial influence on cellular activities in the context of cardiovascular diseases [91]. The TGF-β family comprises three distinct types: TGF-β1, TGF-β2, and TGF-β3, and TGF-β1 is the most extensively researched subtype. Stimulation of TGF-β1 induced VICs to display osteogenic-like characteristics, along with the upregulation of osteogenic genes including Runx2 and Osterix, and a positive correlation with the accumulation of phosphorylated Smad3 in the nucleus [92]. In DAVD, TGF-β1 not only enhances collagen production by VICs but also fosters the equilibrium between MMPs and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), influencing the progression of calcification [93]. The expression of ALP and genes associated with osteogenesis, including osteocalcin, was markedly elevated in VICs exposed to high levels of TGF-β1, indicating an augmentation in calcification deposition [94]. Association of TGF-β1 with inflammatory factors (such as IL-6 and TNF-α) exacerbated the calcification of VICs, and inhibition of the activity of these adenylate acylases significantly reduced the degree of calcification [95].
Furthermore, the Wnt/β-catenin signaling pathway fosters an osteogenic transformation in VICs, resulting in the accumulation of calcium salts and exacerbating the calcification process. When Wnt proteins interact with receptors on the cell membrane, they trigger signal transduction and inhibit GSK3β activity. This leads to the accumulation of β-catenin in the cytoplasm and its subsequent translocation into the nucleus. Within the nucleus, β-catenin binds to transcription factors, activating the expression of numerous genes associated with osteogenesis (Runx2, Osterix and ALP) [96]. The NF-κB signaling pathway is primarily linked to inflammatory responses and immune regulation. When NF-κB is activated, it triggers the release of proinflammatory factors, which subsequently amplify the calcification capabilities of VICs [97]. Therefore, inhibiting NF-κB can diminish the extent of calcification, underscoring its pivotal role in the calcification process. The MAPK signaling pathway is characterized by the activation of p38 MAPK and ERK, which correlates with the osteogenic transformation and calcification of VICs [98]. At the same time, ERK1/2 inhibition-reduced expression of ALP, BMP-2 and Runx2 by activating DKK1 and LRP6 expression could reduce calcification [99].
Other influencing factors
Genetic predisposition
The development of DAVD exhibits a prominent hereditary aspect, evidenced by the clustering of patients afflicted with the condition across multiple generations within extended families in Western France [100]. According to available data, individuals who are siblings of AS patients possess a significantly increased risk of developing AS, exceeding four times that of the general population [101]. In addition to family aggregation, there are genetic polymorphisms, such as certain single-nucleotide polymorphisms (SNPs) in the BMP-2 and BMP-4 genes that are significantly associated with AVC [102]. These findings underscore the significant role of genetics in determining risk, and the potential for identifying genetic markers to offer innovative approaches for therapeutic strategies and enhanced risk assessment.
Metabolism
In the process of DAVD, metabolic disorders pertaining to lipids, glycolipids, calcium, and phosphorus all play a crucial role in the thickening and calcification of cardiac valves. Common metabolic disorders encompass hyperlipidemia, diabetes, and chronic renal insufficiency, which impact valve structure and function via various mechanisms.
Within calcified valve tissue, the presence of inflammatory cell infiltration, lipid deposition, and the formation of foam cells can be observed. This morphological evidence indicates a potential involvement of hyperlipidemia in the pathological processes leading to aortic valve degeneration [103]. Fibroblasts exposed to oxidized LDL (OxLDL) are able to release stromal vesicles and osteocyte-associated proteins that constitute the core of calcification [104]. Downregulation of Sestrin2 (Sesn2) in the Nrf 2 pathway increases the VICs’ osteogenic differentiation induced by OxLDL, regulating the oxidative stress involved in the development of DAVD [105]. The catabolism of oxidized phospholipid (OxPL) into lysophosphatidylcholine (LPC) and free fatty acids triggers activation of the cAMP/PKA signaling cascade, ultimately leading to the transition of VICs into an osteogenic-like phenotype [106]. Lp is the main carrier of OxPL, which is considered to a major contributor to CAVD [107]. Some scholars believe that the combination of the two can predict the risk of AVC in patients with AS [104]. The core protein particle structure of Lp(a) is similar to that of LDL. Since LDL functions as the primary transporter of cholesterol, lowering LDL levels is central to cholesterol-lowering therapy, thereby alleviating valve calcification [108]. In recent years, due to the limited effectiveness of increasing statin doses in reducing LDL and the significantly higher side effects associated with such increases, the possibility of further lipid lowering through high-dose statins has been limited [109]. Cardiologists are beginning to explore new strategies, such as ezetimibe and PCSK9 inhibitors, which can effectively attenuate the progression of mild-to-moderate CAVD in patients by decreasing the concentrations of Lp (a) and LDL [110]. In a secondary analysis from a prospective randomized clinical trial, treatment with simvastatin/ezetimibe combination reduced the need for aortic valve replacement in a subset of patients with mild AS and high pretreatment LDL levels [106]. Proprotein convertase subtilisin/kexin type 9 (PCSK9), a circulating plasma protein that is mainly produced and secreted by the liver, can promote the lysosomal degradation of LDL-R, leading to elevated LDL levels, and PCSK9 inhibitors can lower lipid [111]. Compared with high-intensity statin monotherapy, the combination of statin treatment with ezetimibe or PCSK9 monoclonal antibodies can reduce LDL to 0.8 to 1.4 mmol/L, further decreasing the risk of cardiovascular death, myocardial infarction, and stroke in patients with CAVD [112, 113]. Recent studies have shown that PCSK9 monoclonal antibodies can reduce Lp(a) levels while also lowering LDL, offering a greater cardiovascular protective effect in patients with higher baseline Lp(a) levels. Currently, new small nucleic acid drugs targeting Lp(a) are in the development stage, and clinical trials of these drugs are designed to evaluate their efficacy and safety in reducing Lp(a) levels [114-117].
The impaired insulin sensitivity in patients with type 2 diabetes mellitus is an important trigger for the degenerative process of the cardiovascular system [118]. Retrospective and prospective clinical studies have shown an association between diabetes and an increased risk of severe AS [29, 119]. An environment with high glucose levels can intensify oxidative stress in VICs, exacerbate inflammatory responses, and further lead to the degeneration and calcium deposition of the extracellular matrix. In a study, metformin has demonstrated the ability to alleviate calcification and apoptosis in aortic VICs, thereby presenting a promising therapeutic option for future patients [120]. Advanced glycation end-products (AGEs) are the focus of the current study. Poorly controlled diabetes leads to increased AGEs accumulation in valve, which at least partially, might result in AS progression in diabetes patients [121]. Thus, trying to reduce the accumulation of AGEs in patients with type 2 diabetes may reduce the severity of AS.
An elevated risk cardiovascular calcification is associated with the existence of chronic kidney disease (CKD) [122]. Certain studies have revealed that individuals suffering from DHVD exhibit elevated levels of blood phosphorus, as well as elevated blood calcium phosphorus products. These findings suggest a potential association between this disease and aberrant calcium and phosphorus metabolism. A statistically significant correlation was observed between blood phosphorus levels and the occurrence of AVC, indicating a potential association between phosphorus metabolites in the blood and the development of AVC [123]. Serum alkaline phosphatase levels, which play a crucial role in calcium and phosphorus metabolism, serve as an independent biomarker for AVC incidence and potentially possess the capacity to serve as a predictor of AVC risk factors [124]. Consistently strict phosphate control may slow the progression of coronary and valvular calcifications in patients undergoing hemodialysis [41]. But a latest study showed that the use of vitamin D receptor activators does not reduce the risks of cardiovascular events or all-cause mortality in patients on dialysis with well-controlled secondary hyperparathyroidism [125]. Consequently, ongoing research and exploration are essential to significantly mitigate valve calcification in patients undergoing dialysis.
Moreover, increasing evidence suggests that DAVD could be described as a specific phenotype of heart failure with preserved ejection fraction (HFpEF) [126]. DAVD with preserved ejection fraction and HFpEF display intriguing similarities, especially the inflammatory-metabolic phenotype of the disease is one of the most cardiovascular risk factors, through regulating the inflammatory and atherogenic balance at a local and systemic level by epicardial adipose tissue (EAT) [127, 128]. EAT produces large quantities of anti-inflammatory and pro-inflammatory adipokines through paracrine and vascular secretion, influencing the balance of glucose and lipid metabolism, inflammation and atherogenic factors at both local and systemic levels [129]. Excessive EAT accumulation is associated with hemodynamic disturbances and impaired peripheral oxygen uptake, increasing the risk of major adverse cardiovascular events [130]. The direct association of EAT thickness with more pronounced valve calcifications further supports the relationship between EAT and the severity of DAVD [131].
Hemodynamics and shear stress
With the advancement of DAVD, leaflet flexibility decreased, leading to a significant increase in transaortic jet velocity and hemodynamic impediment. Subsequently, a pressure gradient arises between the LV and the aorta [132]. These localized hemodynamic abnormalities, in turn, can exacerbate the progression of the disease, thus constituting a positive feedback mechanism in biological effects [133]. DAVD is distinguished by the presence of pulsatile shear stress on the ventricular aspect, coupled with low and alternating shear stress on the aortic aspect [134]. Animal model studies have revealed that the application in shear stress is capable of activating the latent TGF-β1, thereby triggering the processes of fibrosis and calcification [135]. Mechanosensitive ion channel Piezol senses shear stress and in turn affects multiple signaling pathways such as BMP/TGFP signaling in aortic valve interstitial cells, leading to changes in cell function and phenotype, gene expression and cell behaviors such as proliferation, migration, apoptosis and remodeling, which subsequently lead to CAVD [136]. An investigation pertaining to the mechanosensitive proteins in valvular cells revealed that the expression levels of CC chemokine ligands, thrombospondin, growth factors, and IL tend to counteract the action of mechanical forces [137]. Aortic valve local turbulent shear can also change and activate several different signal pathways associated with valve calcification stenosis. This leads to the change of the cell cycle, resulting in the release of nitric oxide and prostacyclin, increased oxidative stress response, Lp deposition in the valve of interstitium, inflammatory cells and vascular smooth muscle cell migration, differentiation and hyperplasia, and neoangiogenesis [138]. The role of neoangiogenesis in the development of aortic valve stenosis suggests a potential therapeutic approach in a domain devoid of established effective medical treatments [139].
The bicuspid aortic valve (BAV) malformation is a structure that results from the partial fusion of three aortic leaflets, leading to a valve disease characterized by the presence of only two leaflets instead of the normal three [140]. BAV malformation is the most common congenital heart disease. It is an autosomal dominant condition caused by mutations in chromosomes 18q, 5q, and 13q, with a low population incidence of 1% [141]. It is often accompanied by valve stenosis resulting from dystrophic calcification and progressive dilation of the ascending aorta [142]. Common structural features include variable sizes of leaflets, junction fusion of the leaflets, and irregular morphology. Since the patient’s open valve area is reduced and the size of the leaflets varies, this blocks the flow from the left ventricle into the aorta, causes uneven blood flow distribution, increases flow velocity, and leads to left ventricular hypertrophy. The unstable hemodynamic states produce abnormal shear stress [143, 144], which continuously affects the aortic valve tissue. Over time, the leaflets not only gradually thicken but also exhibit calcification, accompanied by fibrosis and other adverse changes. Ultimately, these alterations can severely impair the heart’s normal systolic and diastolic functions [145].
| Clinical Status | ▴Top |
Diagnosis
Imaging examination plays a crucial role in the diagnosis and evaluation of DAVD. Echocardiography serves as a frequently utilized diagnostic tool for degenerative heart valve disease in the elderly population, affording precise and comprehensive insights into the nature of the lesions [146]. As the gold standard for valvular heart disease, echocardiography not only evaluates the valve morphology and function, but also allows for the quantitative analysis of the degree of valve stenosis. It was observed that the primary pathological alterations observed in DAVD by echocardiography encompass degeneration of the valvular connective tissue, augmentation in leaflet thickness, calcification, and the development of fibrosis. By further measuring the aortic valve orifice area, maximum flow rate, and mean pressure difference, the physician can determine the severity of the aortic valve degeneration [147]. The diagnostic criteria of echocardiography in valvular heart disease adhere strictly to the guidelines issued jointly by the European Association of Cardiovascular Imaging and the American Society of Echocardiography [148]. Furthermore, transesophageal echocardiography offers superior clarity in visualizing structural details, making it ideal for assessing the anatomy in the vicinity of the valve [149]. Since more and more patients with DAVD are asymptomatic at rest, the role of stress echocardiography in the diagnostic workup of DAVD becomes more and more important, which can unveil many relevant hemodynamic and clinical abnormalities [150]. In certain instances, particularly when dealing with complex cases, computed tomography (CT) and nuclear magnetic resonance imaging (MRI) can also be employed to further assess the anatomical structure and function of the valve [151].
Recently, the application of biomarkers has introduced innovative methods for diagnosing DAVD. In particular, proteins associated with heart structure, such as B-type natriuretic peptide (BNP) and its N-terminal precursors (NT-proBNP), are elevated in cases of HF and help physicians assess the cardiac function status of patients [152]. Research has indicated that the concentrations of BNP and NT-proBNP are markedly correlated with the severity of AS [153]. Moreover, biochemical markers associated with MF (markers of collagen metabolism, galectin-3, soluble ST2) and myocyte death/myocardial ischemia (high-sensitivity cardiac troponins, heart-type fatty acid binding protein, myosin-binding protein C) may be future directions of diagnosing biomarkers [154]. Previous studies put forward additional potential biomarkers, including proteins associated with calcification and inflammatory factors, etc. [155, 156].
Treatment progress
Since the drug has no reversal effect on the structural lesions of the aortic valve, patients with DAVD may eventually require surgical intervention. As the sole radical treatment for DAVD, surgical interventions encompass primarily valvuloplasty and valve replacement procedures (Fig. 2) [156]. Presently, a minimally invasive transcatheter aortic valve replacement (TAVR) has emerged as a successful treatment option for severe AS in elderly patients who are unable to endure major surgery [157]. Studies showed comparable safety and efficacy of TAVR in short-term and long-term outcomes compared to traditional surgical procedures, which provides opportunities of surgical intervention to numerous high-risk patients, markedly enhancing their survival rates and quality of life [158]. Especially, TAVR is effective and safe for patients with severe AS, offering results comparable to those of early surgical intervention [159]. Over the past two decades, TAVR has been found to have several limitations. Consequently, there has been a rise in the number of aortic transcatheter heart valves available on the market, aimed at reducing or eliminating adverse outcomes such as paravalvular leakage and conduction barriers that may require permanent pacemaker implantation. These valves are also designed for the long-term durability of biological valves [160]. There were no apparent differences in procedure-related adverse events between patients in the TAVR group and those in the clinical surveillance group who underwent aortic-valve replacement [161].
 Click for large image | Figure 2. Surgically removed degenerative aortic valve specimen. |
With a deeper understanding of the pathogenesis of DAVD, biomarkers possessing specificity are considered potential therapeutic targets. Goody et al authored an outstanding review that succinctly summarized the latest therapeutic strategies proposed for AVC, based on the underlying pathological mechanisms [162]. Di Fusco et al also explored whether medications can prevent or decelerate the progression of DAVD by inhibiting its primary pathophysiological mechanisms [163]. Researchers are investigating cutting-edge approaches, including gene therapy and stem cell therapy, to explore the potential for repairing and regenerating a valve that has already undergone degeneration. A meta-analysis of genome-wide association studies (GWAS) aimed at identifying specific genes [164], or tissue-engineered heart valves (TEHVs) that leverage the multidirectional differentiation potential of stem cells [165], both seek to promote the regeneration of valve cells and slow down the disease process.
Future research direction
Future research of DAVD in the field of pathophysiological mechanisms should be aimed to enhance diagnosis, treatment, patient outcomes and medical resource utilization [166, 167]. The emphasis lies in gaining a profound comprehension of the pathophysiological mechanisms, anatomical structures, and the driving force of the microenvironment, identifying pharmacological interventions aimed at preventing or decelerating the advancement of AS [168, 169]. To enhance the precision of early diagnosis, it is imperative in the future to advance the development of innovative imaging technologies and refine multimodality cardiovascular imaging [170]. Emerging technologies, including optical coherence tomography (OCT) and three-dimensional imaging, have the capability to offer more detailed insights into valve structure and function [171, 172]. Furthermore, the application of genomics and proteomics can also uncover potential biomarkers, facilitating early identification and enabling individualized treatment [173, 174]. There have been studies utilizing clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-engineered isogenic induced pluripotent stem cells for hypertrophic cardiomyopathy. Future research may also investigate the application of techniques like CRISPR to directly repair defective genes in DAVD [175, 176].
| Conclusions | ▴Top |
This review has primarily concentrated on the extant evidence pertaining to the fundamental mechanisms responsible for aortic valve degeneration. The precise mechanisms underlying degenerative heart valvular disease remain enigmatic, and the advancement of novel pharmacological interventions or prophylactic measures for this disease necessitates a more profound comprehension of its pathogenic processes. Comprehensive understanding can identify and develop targeted therapeutic strategies and effectively mitigate the adverse outcomes of this disease. In summary, this review encompasses pathophysiological mechanisms of DADV identified in recent years, indicating the existence of promising therapeutic methods.
Acknowledgments
We would like to express our thanks to the predecessors in this field. Their outstanding achievements are the cornerstone for us to construct this review. We also appreciate the funding support for providing high-quality resources, creating favorable conditions for the review. Once again, we are grateful to all those who have helped us.
Financial Disclosure
This work was supported by Huanhua Talent for Discipline Backbone of Sichuan Provincial People’s Hospital (SY2022017), Science Fund for Distinguished Young Scholars of Sichuan Province (2021JDJQ0041), and National Natural Science and Technology Foundation of China (81800274).
Conflict of Interest
The authors declare no conflict of interest
Author Contributions
Ya Lu Yu and Qin Jiang contributed equally to the manuscript. Ya Lu Yu contributed to drafting the manuscript. Qin Jiang contributed to the discovery of this case and revision of the manuscript. All authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
DAVD: degenerative aortic valve disease; CAVD: calcific aortic valve disease; AS: aortic stenosis; DALYs: disability-adjusted life years; HF: heart failure; VICs: valvular interstitial cells; VECs: valvular endothelial cells; ECM: extracellular matrix; PGs: proteoglycans; GAGs: glycosaminoglycans; TNF-α: tumor necrosis factor-α; IL: interleukin; MMPs: matrix metalloproteinases; EndMT: endothelial-mesenchymal transition; TFs: transcriptional factors; DUSP 1: dual-specificity phosphatase 1; PDK-4: pyruvate dehydrogenase kinase-4; BSP: bone sialoprotein; TGF-β: transforming growth factor β; BMP: bone morphogenetic protein; miRNA: microRNA; MF: myocardial fibrosis; LA: left atrium; lncRNA: long non-coding RNA; AVC: aortic valve calcification; AF: atrial fibrillation; α-SMA: α-smooth muscle actin; eNOS: endothelial nitric oxide synthase; NO: nitric oxide; NOS3: nitric oxide synthase 3; Runx2: Runt-related transcription factor; ICAM-1: intercellular adhesion molecule-1; LPS: lipopolysaccharide; PG: peptidoglycan; TIMPs: tissue inhibitors of metalloproteinases; SNPs: single-nucleotide polymorphisms; OxLDL: oxidized LDL; Sesn2: sestrin2; OxPL: oxidized phospholipid; Lp: lipoprotein; LPC: lysophosphatidylcholine; AGEs: advanced glycation end-products; CKD: chronic kidney disease; HFpEF: heart failure with preserved ejection fraction; EAT: epicardial adipose tissue; BAV: bicuspid aortic valve; CT: computed tomography; MRI: magnetic resonance imaging; TAVR: transcatheter aortic valve replacement; GWAS: genome-wide association studies; TEHVs: tissue-engineered heart valves; OCT: optical coherence tomography
| References | ▴Top |
- Yang Y, Wang Z, Chen Z, Wang X, Zhang L, Li S, Zheng C, et al. Current status and etiology of valvular heart disease in China: a population-based survey. BMC Cardiovasc Disord. 2021;21(1):339.
doi pubmed - Pinto G, Fragasso G. Aortic valve stenosis: drivers of disease progression and drug targets for therapeutic opportunities. Expert Opin Ther Targets. 2022;26(7):633-644.
doi pubmed - Abecasis J, Gomes Pinto D, Ramos S, Masci PG, Cardim N, Gil V, Felix A. Left ventricular remodeling in degenerative aortic valve stenosis. Curr Probl Cardiol. 2021;46(5):100801.
doi pubmed - Lerman DA, Prasad S, Alotti N. Calcific aortic valve disease: molecular mechanisms and therapeutic approaches. Eur Cardiol. 2015;10(2):108-112.
doi pubmed - Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124(16):1783-1791.
doi pubmed - Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of 'degenerative' valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90(2):844-853.
doi pubmed - Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113(2):198-208.
doi pubmed - Oury C, Nchimi A, Lancellotti P. Editorial: from biology to clinical management: an update on aortic valve disease. Front Cardiovasc Med. 2019;6:4.
doi pubmed - Yadgir S, Johnson CO, Aboyans V, Adebayo OM, Adedoyin RA, Afarideh M, Alahdab F, et al. Global, regional, and national burden of calcific aortic valve and degenerative mitral valve diseases, 1990-2017. Circulation. 2020;141(21):1670-1680.
doi pubmed - Coffey S, Roberts-Thomson R, Brown A, Carapetis J, Chen M, Enriquez-Sarano M, Zuhlke L, et al. Global epidemiology of valvular heart disease. Nat Rev Cardiol. 2021;18(12):853-864.
doi pubmed - Iung B, Baron G, Butchart EG, Delahaye F, Gohlke-Barwolf C, Levang OW, Tornos P, et al. A prospective survey of patients with valvular heart disease in Europe: The Euro Heart Survey on Valvular Heart Disease. Eur Heart J. 2003;24(13):1231-1243.
doi pubmed - Tzeng H, et al. Prevalence of asymptomatic severe aortic stenosis in a community-based elderly population. Am J Cardiol. 2006;98(7):981-984.
- Di Vito A, Donato A, Presta I, Mancuso T, Brunetti FS, Mastroroberto P, Amorosi A, et al. Extracellular matrix in calcific aortic valve disease: architecture, dynamic and perspectives. Int J Mol Sci. 2021;22(2):913.
doi pubmed - DesJardin JT, Chikwe J, Hahn RT, Hung JW, Delling FN. Sex differences and similarities in valvular heart disease. Circ Res. 2022;130(4):455-473.
doi pubmed - Leibowitz D, Yoshida Y, Jin Z, Nakanishi K, Mannina C, Elkind MSV, Rundek T, et al. Factors associated with the progression of aortic valve calcification in older adults. Int J Cardiol. 2023;381:76-80.
doi pubmed - Kaiser Y, van der Toorn JE, Singh SS, Zheng KH, Kavousi M, Sijbrands EJG, Stroes ESG, et al. Lipoprotein(a) is associated with the onset but not the progression of aortic valve calcification. Eur Heart J. 2022;43(39):3960-3967.
doi pubmed - Rodriguez-Padial L, Moreu Burgos J, Barderas MG. Coronary flow reserve in degenerative aortic stenosis and diabetes mellitus: An intriguing question. Kardiol Pol. 2022;80(12):1187-1189.
doi pubmed - Duran CM, et al. Valvular heart disease and heart failure in the elderly: facts and misconceptions. Heart Fail Clin. 2011;7(2):221-231.
- Freedman RA, et al. Atrial fibrillation and its relationship to valvular heart disease. Heart. 2011;97(22):1896-1900.
- Zhang S, Liu C, Wu P, Li H, Zhang Y, Feng K, Huang H, et al. Burden and temporal trends of valvular heart disease-related heart failure from 1990 to 2019 and projection up to 2030 in group of 20 countries: an analysis for the global burden of disease study 2019. J Am Heart Assoc. 2024;13(20):e036462.
doi pubmed - Lansakara M, Unai S. An overview of aortic valve anatomy: the current understanding. Indian J Thorac Cardiovasc Surg. 2023;39(Suppl 2):246-252.
doi pubmed - Perrucci GL, Zanobini M, Gripari P, Songia P, Alshaikh B, Tremoli E, Poggio P. Pathophysiology of aortic stenosis and mitral regurgitation. Compr Physiol. 2017;7(3):799-818.
doi pubmed - Kodigepalli KM, Thatcher K, West T, Howsmon DP, Schoen FJ, Sacks MS, Breuer CK, et al. Biology and biomechanics of the heart valve extracellular matrix. J Cardiovasc Dev Dis. 2020;7(4):57.
doi pubmed - Sohmer B, Jafar R, Patel P, Chamberland ME, Labrosse MR, Boodhwani M. Aortic valve cusp coaptation surface area using 3-dimensional transesophageal echocardiography correlates with severity of aortic valve insufficiency. J Cardiothorac Vasc Anesth. 2018;32(1):344-351.
doi pubmed - Chester AH, El-Hamamsy I, Butcher JT, Latif N, Bertazzo S, Yacoub MH. The living aortic valve: From molecules to function. Glob Cardiol Sci Pract. 2014;2014(1):52-77.
doi pubmed - Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15(8):445-456.
doi pubmed - Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD, Lincoln J. Valve Endothelial cell-derived Tgfbeta1 signaling promotes nuclear localization of Sox9 in interstitial cells associated with attenuated calcification. Arterioscler Thromb Vasc Biol. 2016;36(2):328-338.
doi pubmed - Gould ST, Matherly EE, Smith JN, Heistad DD, Anseth KS. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials. 2014;35(11):3596-3606.
doi pubmed - Selig JI, Ouwens DM, Raschke S, Thoresen GH, Fischer JW, Lichtenberg A, Akhyari P, et al. Impact of hyperinsulinemia and hyperglycemia on valvular interstitial cells - A link between aortic heart valve degeneration and type 2 diabetes. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2526-2537.
doi pubmed - Pierlot CM, Lee JM, Amini R, Sacks MS, Wells SM. Pregnancy-induced remodeling of collagen architecture and content in the mitral valve. Ann Biomed Eng. 2014;42(10):2058-2071.
doi pubmed - Yip CY, Simmons CA. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovasc Pathol. 2011;20(3):177-182.
doi pubmed - Falla Zuniga LF, Munoz Ceron YS, Salazar L. Structural remodelling of the heart valves extracellular matrix during embryo development. Anat Histol Embryol. 2021;50(1):206-211.
doi pubmed - Yu Chen H, Dina C, Small AM, Shaffer CM, Levinson RT, Helgadottir A, Capoulade R, et al. Dyslipidemia, inflammation, calcification, and adiposity in aortic stenosis: a genome-wide study. Eur Heart J. 2023;44(21):1927-1939.
doi pubmed - Jiang Q, Yu T, Huang K, Zhang H, Zheng Z, Hu S. Systemic redistribution of the intramyocardially injected mesenchymal stem cells by repeated remote ischaemic post-conditioning. J Cell Mol Med. 2018;22(1):417-428.
doi pubmed - Blake RR, Markby GR, Culshaw GJ, Martinez-Pereira Y, Lu CC, Corcoran BM. Survival of activated myofibroblasts in canine myxomatous mitral valve disease and the role of apoptosis. Res Vet Sci. 2020;128:99-106.
doi pubmed - Galli D, Manuguerra R, Monaco R, Manotti L, Goldoni M, Becchi G, Carubbi C, et al. Understanding the structural features of symptomatic calcific aortic valve stenosis: A broad-spectrum clinico-pathologic study in 236 consecutive surgical cases. Int J Cardiol. 2017;228:364-374.
doi pubmed - Conte M, Petraglia L, Campana P, Gerundo G, Caruso A, Grimaldi MG, Russo V, et al. The role of inflammation and metabolic risk factors in the pathogenesis of calcific aortic valve stenosis. Aging Clin Exp Res. 2021;33(7):1765-1770.
doi pubmed - Combi Z, Potor L, Nagy P, Sikura KE, Ditroi T, Juranyi EP, Galambos K, et al. Hydrogen sulfide as an anti-calcification stratagem in human aortic valve: Altered biogenesis and mitochondrial metabolism of H(2)S lead to H(2)S deficiency in calcific aortic valve disease. Redox Biol. 2023;60:102629.
doi pubmed - Shu Y, Jin S. Caveolin-1 in endothelial cells: A potential therapeutic target for atherosclerosis. Heliyon. 2023;9(8):e18653.
doi pubmed - Gong HJ, Lin JJ, Li H, Nan ZQ. A study on protective effect of morphine against myocardial ischemia-reperfusion injury in rats via CAMP/PKA signaling pathway. J Biol Regul Homeost Agents. 2020;34(5):1669-1677.
doi pubmed - Gonzalez Rodriguez A, Schroeder ME, Grim JC, Walker CJ, Speckl KF, Weiss RM, Anseth KS. Tumor necrosis factor-alpha promotes and exacerbates calcification in heart valve myofibroblast populations. FASEB J. 2021;35(3):e21382.
doi pubmed - Yang Z, Zhang J, Zhu Y, Zhang C, Li G, Liu S, Du J, et al. IL-17A induces valvular endothelial inflammation and aggravates calcific aortic valve disease. Biochem Biophys Res Commun. 2023;672:145-153.
doi pubmed - The E, Zhai Y, Yao Q, Ao L, Li S, Fullerton DA, Dinarello CA, et al. Recombinant IL-37 exerts an anti-inflammatory effect on human aortic valve interstitial cells through extracellular and intracellular actions. Int J Biol Sci. 2023;19(12):3908-3919.
doi pubmed - Candellier A, Issa N, Grissi M, Brouette T, Avondo C, Gomila C, Blot G, et al. Indoxyl-sulfate activation of the AhR- NF-kappaB pathway promotes interleukin-6 secretion and the subsequent osteogenic differentiation of human valvular interstitial cells from the aortic valve. J Mol Cell Cardiol. 2023;179:18-29.
doi pubmed - Zickler D, Luecht C, Willy K, Chen L, Witowski J, Girndt M, Fiedler R, et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol Dial Transplant. 2018;33(4):574-585.
doi pubmed - Dayawansa NH, Baratchi S, Peter K. Uncoupling the vicious cycle of mechanical stress and inflammation in calcific aortic valve disease. Front Cardiovasc Med. 2022;9:783543.
doi pubmed - Walker CJ, Batan D, Bishop CT, Ramirez D, Aguado BA, Schroeder ME, Crocini C, et al. Extracellular matrix stiffness controls cardiac valve myofibroblast activation through epigenetic remodeling. Bioeng Transl Med. 2022;7(3):e10394.
doi pubmed - Pasipoularides A. Calcific Aortic Valve Disease: Part 1—molecular pathogenetic aspects, hemodynamics, and adaptive feedbacks. J Cardiovasc Transl Res. 2016;9(2):102-118.
doi pubmed - Akahori H, Tsujino T, Masuyama T, Ishihara M. Mechanisms of aortic stenosis. J Cardiol. 2018;71(3):215-220.
doi pubmed - Yu C, Wu D, Zhao C, Wu C. Overexpressed thrombospondin 2 induced osteogenic differentiation of valve interstitial cells via inhibition of Akt/NF-kappaB signaling pathway to promote calcific aortic valve disease development. Dis Markers. 2022;2022:2022958.
doi pubmed - Jung JJ, Razavian M, Kim HY, Ye Y, Golestani R, Toczek J, Zhang J, et al. Matrix metalloproteinase inhibitor, doxycycline and progression of calcific aortic valve disease in hyperlipidemic mice. Sci Rep. 2016;6:32659.
doi pubmed - Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun. 2017;8:14361.
doi pubmed - Peng Q, Shan D, Cui K, Li K, Zhu B, Wu H, Wang B, et al. The role of endothelial-to-mesenchymal transition in cardiovascular disease. Cells. 2022;11(11):1834.
doi pubmed - Li A, Peng W, Xia X, Li R, Wang Y, Wei D. Endothelial-to-mesenchymal transition: a potential mechanism for atherosclerosis plaque progression and destabilization. DNA Cell Biol. 2017;36(11):883-891.
doi pubmed - Gorelova A, Berman M, Al Ghouleh I. Endothelial-to-mesenchymal transition in pulmonary arterial hypertension. Antioxid Redox Signal. 2021;34(12):891-914.
doi pubmed - Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021;41(9):2357-2369.
doi pubmed - Wang Y, Xiao X, Zhou T, Han D, Dong N. Novel mechanisms for osteogenic differentiation of human aortic valve interstitial cells. J Thorac Cardiovasc Surg. 2020;159(5):1742-1753.e7.
doi pubmed - Anger T, Carson W, Weyand M, Daniel WG, Hoeher M, Garlichs CD. Atherosclerotic inflammation triggers osteogenic bone transformation in calcified and stenotic human aortic valves: still a matter of debate. Exp Mol Pathol. 2009;86(1):10-17.
doi pubmed - Zhou T, Han D, Liu J, Shi J, Zhu P, Wang Y, Dong N. Factors influencing osteogenic differentiation of human aortic valve interstitial cells. J Thorac Cardiovasc Surg. 2021;161(2):e163-e185.
doi pubmed - Huang Y, Jiang C, Chen L, Han J, Liu M, Zhou T, Dong N, et al. Gli1 promotes the phenotypic transformation of valve interstitial cells through Hedgehog pathway activation exacerbating calcific aortic valve disease. Int J Biol Sci. 2023;19(7):2053-2066.
doi pubmed - Fernandez-Pisonero I, Lopez J, Onecha E, Duenas AI, Maeso P, Crespo MS, San Roman JA, et al. Synergy between sphingosine 1-phosphate and lipopolysaccharide signaling promotes an inflammatory, angiogenic and osteogenic response in human aortic valve interstitial cells. PLoS One. 2014;9(9):e109081.
doi pubmed - Jiang Q, Huang K, Wang D, Xia J, Yu T, Hu S. A comparison of bilateral and unilateral cerebral perfusion for total arch replacement surgery for non-marfan, type A aortic dissection. Perfusion. 2024;39(6):1070-1079.
doi pubmed - Xu HX, Wang Y, Zheng DD, Wang T, Pan M, Shi JH, Zhu JH, et al. Differential expression of MicroRNAs in calcific aortic stenosis. Clin Lab. 2017;63(7):1163-1170.
doi pubmed - Xiao X, Zhou T, Guo S, Guo C, Zhang Q, Dong N, Wang Y. LncRNA MALAT1 sponges miR-204 to promote osteoblast differentiation of human aortic valve interstitial cells through up-regulating Smad4. Int J Cardiol. 2017;243:404-412.
doi pubmed - Zheng Y, Wen S, Jiang S, He S, Qiao W, Liu Y, Yang W, et al. CircRNA/lncRNA-miRNA-mRNA network and gene landscape in calcific aortic valve disease. BMC Genomics. 2023;24(1):419.
doi pubmed - Wang W, Jiang Q, Zhang H, Jin P, Yuan X, Wei Y, Hu S. Intravenous administration of bone marrow mesenchymal stromal cells is safe for the lung in a chronic myocardial infarction model. Regen Med. 2011;6(2):179-190.
doi pubmed - Jiang Q, Xiang B, Wang H, Huang K, Kong H, Hu S. Remote ischaemic preconditioning ameliorates sinus rhythm restoration rate through Cox maze radiofrequency procedure associated with inflammation reaction reduction. Basic Res Cardiol. 2019;114(3):14.
doi pubmed - Jiang Q, Yu T, Huang K, Lu J, Zhang H, Hu S. Remote ischemic postconditioning ameliorates the mesenchymal stem cells engraftment in reperfused myocardium. PLoS One. 2016;11(1):e0146074.
doi pubmed - Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, Ghaffari SH. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J Cell Physiol. 2019;234(5):5451-5465.
doi pubmed - Xu R, Zhao M, Yang Y, Huang Z, Shi C, Hou X, Zhao Y, et al. MicroRNA-449c-5p inhibits osteogenic differentiation of human VICs through Smad4-mediated pathway. Sci Rep. 2017;7(1):8740.
doi pubmed - Fabiani I, Scatena C, Mazzanti CM, Conte L, Pugliese NR, Franceschi S, Lessi F, et al. Micro-RNA-21 (biomarker) and global longitudinal strain (functional marker) in detection of myocardial fibrotic burden in severe aortic valve stenosis: a pilot study. J Transl Med. 2016;14(1):248.
doi pubmed - Ohukainen P, Syvaranta S, Napankangas J, Rajamaki K, Taskinen P, Peltonen T, Helske-Suihko S, et al. MicroRNA-125b and chemokine CCL4 expression are associated with calcific aortic valve disease. Ann Med. 2015;47(5):423-429.
doi pubmed - Yan F, Huo Q, Zhang W, Wu T, Dilimulati D, Shi L. MiR-138-5p targets RUNX2 to inhibit osteogenic differentiation of aortic valve interstitial cells via Wnt/beta-catenin signaling pathway. BMC Cardiovasc Disord. 2022;22(1):24.
doi pubmed - Salim MT, Esmerats JF, Arjunon S, Villa-Roel N, Nerem RM, Jo H, Yoganathan AP. miR-214 is stretch-sensitive in aortic valve and inhibits aortic valve calcification. Ann Biomed Eng. 2019;47(4):1106-1115.
doi pubmed - Zhou H, Lin S, Li X, Guo D, Wang Y, Hu Y. Serum miR-222 is independently associated with atrial fibrillation in patients with degenerative valvular heart disease. BMC Cardiovasc Disord. 2021;21(1):98.
doi pubmed - Tan J, Hu L, Yang X, Zhang X, Wei C, Lu Q, Chen Z, et al. miRNA expression profiling uncovers a role of miR-302b-3p in regulating skin fibroblasts senescence. J Cell Biochem. 2020;121(1):70-80.
doi pubmed - Jiang Q, Song P, Wang E, Li J, Hu S, Zhang H. Remote ischemic postconditioning enhances cell retention in the myocardium after intravenous administration of bone marrow mesenchymal stromal cells. J Mol Cell Cardiol. 2013;56:1-7.
doi pubmed - Qin JZ, Wang SJ, Xia C. microRNAs regulate nitric oxide release from endothelial cells by targeting NOS3. J Thromb Thrombolysis. 2018;46(3):275-282.
doi pubmed - Salviano-Silva A, Lobo-Alves SC, Almeida RC, Malheiros D, Petzl-Erler ML. Besides pathology: long non-coding RNA in cell and tissue homeostasis. Noncoding RNA. 2018;4(1):3.
doi pubmed - Song GY, Guo XN, Yao J, Lu ZN, Xie JH, Wu F, He J, et al. Differential expression profiles and functional analysis of long non-coding RNAs in calcific aortic valve disease. BMC Cardiovasc Disord. 2023;23(1):326.
doi pubmed - He W, Li F, Zhang S, Zhu Z, Lin M, Ge S, Zhou R. LncRNA AFAP1-AS1 promotes osteoblast differentiation of human aortic valve interstitial cells through regulating miR-155/SMAD5 axis. Mol Cell Probes. 2020;50:101509.
doi pubmed - He W, Che H, Jin C, Li Y, Li F, Zhou R. LncRNA AFAP1-AS1 promotes M1 polarization of macrophages and osteogenic differentiation of valve interstitial cells. J Physiol Biochem. 2021;77(3):461-468.
doi pubmed - Zheng D, Wang B, Zhu X, Hu J, Sun J, Xuan J, Ge Z. LncRNA OIP5-AS1 inhibits osteoblast differentiation of valve interstitial cells via miR-137/TWIST11 axis. Biochem Biophys Res Commun. 2019;511(4):826-832.
doi pubmed - Yang R, Tang Y, Chen X, Yang Y. Telocytes-derived extracellular vesicles alleviate aortic valve calcification by carrying miR-30b. ESC Heart Fail. 2021;8(5):3935-3946.
doi pubmed - Sahoo BR. Structure of fish Toll-like receptors (TLR) and NOD-like receptors (NLR). Int J Biol Macromol. 2020;161:1602-1617.
doi pubmed - Zeng Q, Song R, Ao L, Weyant MJ, Lee J, Xu D, Fullerton DA, et al. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-kappaB activation. Arterioscler Thromb Vasc Biol. 2013;33(7):1580-1590.
doi pubmed - Wang Y, Fang Y, Lu P, Wu B, Zhou B. NOTCH signaling in aortic valve development and calcific aortic valve disease. Front Cardiovasc Med. 2021;8:682298.
doi pubmed - Wagley Y, Chesi A, Acevedo PK, Lu S, Wells AD, Johnson ME, Grant SFA, et al. Canonical Notch signaling is required for bone morphogenetic protein-mediated human osteoblast differentiation. Stem Cells. 2020;38(10):1332-1347.
doi pubmed - Zeng P, Yang J, Liu L, Yang X, Yao Z, Ma C, Zhu H, et al. ERK1/2 inhibition reduces vascular calcification by activating miR-126-3p-DKK1/LRP6 pathway. Theranostics. 2021;11(3):1129-1146.
doi pubmed - Luo W, Song Y, Wang J, Yang X, Li Z, Cong H. The effect of osteoprotectin (OPG)/receptor activator of nuclear factor-kappaB Ligand (RANKL)/receptor activator of nuclear factor-kappaB (RANK) gene methylation on aortic valve calcified. Biomed Res Int. 2022;2022:1592576.
doi pubmed - Podolec J, Baran J, Siedlinski M, Urbanczyk M, Krupinski M, Bartus K, Niewiara L, et al. Serum rantes, transforming growth factor-beta1 and interleukin-6 levels correlate with cardiac muscle fibrosis in patients with aortic valve stenosis. J Physiol Pharmacol. 2018;69(4):615-623.
doi pubmed - Song R, Fullerton DA, Ao L, Zheng D, Zhao KS, Meng X. BMP-2 and TGF-beta1 mediate biglycan-induced pro-osteogenic reprogramming in aortic valve interstitial cells. J Mol Med (Berl). 2015;93(4):403-412.
doi pubmed - Jenke A, Kistner J, Saradar S, Chekhoeva A, Yazdanyar M, Bergmann AK, Rotepohl MV, et al. Transforming growth factor-beta1 promotes fibrosis but attenuates calcification of valvular tissue applied as a three-dimensional calcific aortic valve disease model. Am J Physiol Heart Circ Physiol. 2020;319(5):H1123-H1141.
doi pubmed - Zhang C, Liu M, Wang X, Chen S, Fu X, Li G, Dong N, et al. Mechanism of CircANKRD36 regulating cell heterogeneity and endothelial mesenchymal transition in aortic valve stromal cells by regulating miR-599 and TGF-beta signaling pathway. Int J Cardiol. 2022;352:104-114.
doi pubmed - Palafox-Mariscal LA, Ortiz-Lazareno PC, Jave-Suarez LF, Aguilar-Lemarroy A, Villasenor-Garcia MM, Cruz-Lozano JR, Gonzalez-Martinez KL, et al. Pentoxifylline inhibits TNF-alpha/TGF-beta1-Induced Epithelial-Mesenchymal transition via suppressing the NF-kappaB pathway and SERPINE1 expression in CaSki cells. Int J Mol Sci. 2023;24(13):10592.
doi pubmed - Gu GJ, Chen T, Zhou HM, Sun KX, Li J. Role of Wnt/beta-catenin signaling pathway in the mechanism of calcification of aortic valve. J Huazhong Univ Sci Technolog Med Sci. 2014;34(1):33-36.
doi pubmed - Wang D, Zeng Q, Song R, Ao L, Fullerton DA, Meng X. Ligation of ICAM-1 on human aortic valve interstitial cells induces the osteogenic response: A critical role of the Notch1-NF-kappaB pathway in BMP-2 expression. Biochim Biophys Acta. 2014;1843(11):2744-2753.
doi pubmed - Xie F, Li F, Li R, Liu Z, Shi J, Zhang C, Dong N. Inhibition of PP2A enhances the osteogenic differentiation of human aortic valvular interstitial cells via ERK and p38 MAPK pathways. Life Sci. 2020;257:118086.
doi pubmed - Rattazzi M, Faggin E, Bertacco E, Buso R, Puato M, Plebani M, Zaninotto M, et al. RANKL expression is increased in circulating mononuclear cells of patients with calcific aortic stenosis. J Cardiovasc Transl Res. 2018;11(4):329-338.
doi pubmed - Probst V, Le Scouarnec S, Legendre A, Jousseaume V, Jaafar P, Nguyen JM, Chaventre A, et al. Familial aggregation of calcific aortic valve stenosis in the western part of France. Circulation. 2006;113(6):856-860.
doi pubmed - Martinsson A, Li X, Zoller B, Andell P, Andersson C, Sundquist K, Smith JG. Familial aggregation of aortic valvular stenosis: a nationwide study of sibling risk. Circ Cardiovasc Genet. 2017;10(6):e001742.
doi pubmed - Li FF, Deng X, Zhou J, Yan P, Zhao EY, Liu SL. Characterization of human bone morphogenetic protein gene variants for possible roles in congenital heart disease. Mol Med Rep. 2016;14(2):1459-1464.
doi pubmed - Wang H, Xi J, Zhang Z, Li J, Guo L, Li N, Sun Y, et al. Sestrin2 is increased in calcific aortic disease and inhibits osteoblastic differentiation in valvular interstitial cells via the nuclear factor E2-related factor 2 pathway. J Cardiovasc Pharmacol. 2022;80(4):609-615.
doi pubmed - Ozkan U, Ozcelik F, Yildiz M, Budak M. Lipoprotein(a) gene polymorphism increases a risk factor for aortic valve calcification. J Cardiovasc Dev Dis. 2019;6(3):31.
doi pubmed - Capoulade R, Torzewski M, Mayr M, Chan KL, Mathieu P, Bosse Y, Dumesnil JG, et al. ApoCIII-Lp(a) complexes in conjunction with Lp(a)-OxPL predict rapid progression of aortic stenosis. Heart. 2020;106(10):738-745.
doi pubmed - Greve AM, Bang CN, Boman K, Egstrup K, Kesaniemi YA, Ray S, Pedersen TR, et al. Relation of lipid-lowering therapy to need for aortic valve replacement in patients with asymptomatic mild to moderate aortic stenosis. Am J Cardiol. 2019;124(11):1736-1740.
doi pubmed - Yang Y, Hong Y, Yang W, Zheng Z. Association of lipoprotein(a) with aortic dissection. Clin Cardiol. 2022;45(9):908-912.
doi pubmed - Nozue T, Kawashiri MA, Higashikata T, Nohara A, Inazu A, Kobayashi J, Koizumi J, et al. Cholesterol-years score is associated with development of senile degenerative aortic stenosis in heterozygous familial hypercholesterolemia. J Atheroscler Thromb. 2006;13(6):323-328.
doi pubmed - Lee TC, Leung WC, Ho C, Chiu MW, Leung IY, Wong YK, Roxanna LK, et al. Association of LDL-cholesterol <1.8 mmol/L and statin use with the recurrence of intracerebral hemorrhage. Int J Stroke. 2024;19(6):695-704.
doi pubmed - Burger PM, Dorresteijn JAN, Koudstaal S, Holtrop J, Kastelein JJP, Jukema JW, Ridker PM, et al. Course of the effects of LDL-cholesterol reduction on cardiovascular risk over time: A meta-analysis of 60 randomized controlled trials. Atherosclerosis. 2024;396:118540.
doi pubmed - Murphy SA, Pedersen TR, Gaciong ZA, Ceska R, Ezhov MV, Connolly DL, Jukema JW, et al. Effect of the PCSK9 inhibitor evolocumab on total cardiovascular events in patients with cardiovascular disease: a prespecified analysis from the FOURIER trial. JAMA Cardiol. 2019;4(7):613-619.
doi pubmed - Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722.
doi pubmed - Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097-2107.
doi pubmed - Tsimikas S, Karwatowska-Prokopczuk E, Xia S. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. Reply. N Engl J Med. 2020;382(21):e65.
doi pubmed - O'Donoghue ML, Rosenson RS, Gencer B, Lopez JAG, Lepor NE, Baum SJ, Stout E, et al. Small interfering RNA to reduce lipoprotein(a) in cardiovascular disease. N Engl J Med. 2022;387(20):1855-1864.
doi pubmed - Nissen SE, Linnebjerg H, Shen X, Wolski K, Ma X, Lim S, Michael LF, et al. Lepodisiran, an extended-duration short interfering RNA targeting lipoprotein(a): a randomized dose-ascending clinical trial. JAMA. 2023;330(21):2075-2083.
doi pubmed - Nissen SE, Wang Q, Nicholls SJ, Navar AM, Ray KK, Schwartz GG, Szarek M, et al. Zerlasiran-a small-interfering RNA targeting lipoprotein(a): a phase 2 randomized clinical trial. JAMA. 2024;332(23):1992-2002.
doi pubmed - Larsson SC, Wallin A, Hakansson N, Stackelberg O, Back M, Wolk A. Type 1 and type 2 diabetes mellitus and incidence of seven cardiovascular diseases. Int J Cardiol. 2018;262:66-70.
doi pubmed - Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6(1):a009191.
doi pubmed - Deacon CF. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2020;16(11):642-653.
doi pubmed - Hutcheson JD, Goettsch C. Cardiovascular calcification heterogeneity in chronic kidney disease. Circ Res. 2023;132(8):993-1012.
doi pubmed - Linefsky JP, O'Brien KD, Sachs M, Katz R, Eng J, Michos ED, Budoff MJ, et al. Serum phosphate is associated with aortic valve calcification in the Multi-ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2014;233(2):331-337.
doi pubmed - Shimizu M, Fujii H, Kono K, Goto S, Watanabe K, Sakamoto K, Nishi S. Clinical implication of consistently strict phosphate control for coronary and valvular calcification in incident patients undergoing hemodialysis. J Atheroscler Thromb. 2023;30(11):1568-1579.
doi pubmed - Yoshida K, Mizukami T, Fukagawa M, Akizawa T, Morohoshi H, Sambe T, Takeshima A, et al. Effect of vitamin D receptor activators on cardiovascular events in patients on hemodialysis-A post hoc analysis of the LANDMARK study. Ther Apher Dial. 2023;27(3):523-529.
doi pubmed - Wang S, Tong M, Hu S, Chen X. The bioactive substance secreted by MSC retards mouse aortic vascular smooth muscle cells calcification. Biomed Res Int. 2018;2018:6053567.
doi pubmed - De Biase N, Mazzola M, Del Punta L, Di Fiore V, De Carlo M, Giannini C, Costa G, et al. Haemodynamic and metabolic phenotyping of patients with aortic stenosis and preserved ejection fraction: A specific phenotype of heart failure with preserved ejection fraction? Eur J Heart Fail. 2023;25(11):1947-1958.
doi pubmed - Packer M, Lam CSP, Lund LH, Maurer MS, Borlaug BA. Characterization of the inflammatory-metabolic phenotype of heart failure with a preserved ejection fraction: a hypothesis to explain influence of sex on the evolution and potential treatment of the disease. Eur J Heart Fail. 2020;22(9):1551-1567.
doi pubmed - Iacobellis G. Epicardial adipose tissue in contemporary cardiology. Nat Rev Cardiol. 2022;19(9):593-606.
doi pubmed - Pugliese NR, Paneni F, Mazzola M, De Biase N, Del Punta L, Gargani L, Mengozzi A, et al. Impact of epicardial adipose tissue on cardiovascular haemodynamics, metabolic profile, and prognosis in heart failure. Eur J Heart Fail. 2021;23(11):1858-1871.
doi pubmed - Koepp KE, Obokata M, Reddy YNV, Olson TP, Borlaug BA. Hemodynamic and functional impact of epicardial adipose tissue in heart failure with preserved ejection fraction. JACC Heart Fail. 2020;8(8):657-666.
doi pubmed - Pugliese NR, Pellicori P, Filidei F, De Biase N, Maffia P, Guzik TJ, Masi S, et al. Inflammatory pathways in heart failure with preserved left ventricular ejection fraction: implications for future interventions. Cardiovasc Res. 2023;118(18):3536-3555.
doi pubmed - Siasos G, Tsigkou V, Coskun AU, Oikonomou E, Zaromitidou M, Lerman LO, Lerman A, et al. The role of shear stress in coronary artery disease. Curr Top Med Chem. 2023;23(22):2132-2157.
doi pubmed - Ali MS, Wang X, Lacerda CM. A survey of membrane receptor regulation in valvular interstitial cells cultured under mechanical stresses. Exp Cell Res. 2017;351(2):150-156.
doi pubmed - Ahamed J, Burg N, Yoshinaga K, Janczak CA, Rifkin DB, Coller BS. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood. 2008;112(9):3650-3660.
doi pubmed - Fernandez Esmerats J, Heath J, Jo H. Shear-sensitive genes in aortic valve endothelium. Antioxid Redox Signal. 2016;25(7):401-414.
doi pubmed - Heath JM, Fernandez Esmerats J, Khambouneheuang L, Kumar S, Simmons R, Jo H. Mechanosensitive microRNA-181b regulates aortic valve endothelial matrix degradation by targeting TIMP3. Cardiovasc Eng Technol. 2018;9(2):141-150.
doi pubmed - Sun L, Chandra S, Sucosky P. Ex vivo evidence for the contribution of hemodynamic shear stress abnormalities to the early pathogenesis of calcific bicuspid aortic valve disease. PLoS One. 2012;7(10):e48843.
doi pubmed - de Oliveira Sa MPB, Cavalcanti LRP, Perazzo AM, Gomes RAF, Clavel MA, Pibarot P, Biondi-Zoccai G, et al. Calcific aortic valve stenosis and atherosclerotic calcification. Curr Atheroscler Rep. 2020;22(2):2.
doi pubmed - Cho KI, Sakuma I, Sohn IS, Jo SH, Koh KK. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis. 2018;277:60-65.
doi pubmed - Verma R, Cohen G, Colbert J, Fedak PWM. Bicuspid aortic valve associated aortopathy: 2022 guideline update. Curr Opin Cardiol. 2023;38(2):61-67.
doi pubmed - Martin LJ, Ramachandran V, Cripe LH, Hinton RB, Andelfinger G, Tabangin M, Shooner K, et al. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum Genet. 2007;121(2):275-284.
doi pubmed - Thiene G, Rizzo S, Basso C. Bicuspid aortic valve: The most frequent and not so benign congenital heart disease. Cardiovasc Pathol. 2024;70:107604.
doi pubmed - Berdajs D, Mosbahi S, Eckstein FS, Charbonnier D, Ferrari E, von Segesser LK. Impact of the bicuspid aortic valve on aortic root haemodynamics: three-dimensional computed fluid dynamics simulation. Interact Cardiovasc Thorac Surg. 2018;27(3):446-454.
doi pubmed - Guala A, Evangelista A, Teixido-Tura G, La Mura L, Dux-Santoy L, Ruiz-Munoz A, Valente F, et al. Leaflet fusion length is associated with aortic dilation and flow alterations in non-dysfunctional bicuspid aortic valve. Eur Radiol. 2021;31(12):9262-9272.
doi pubmed - Chandra S, Rajamannan NM, Sucosky P. Computational assessment of bicuspid aortic valve wall-shear stress: implications for calcific aortic valve disease. Biomech Model Mechanobiol. 2012;11(7):1085-1096.
doi pubmed - Nagueh SF, Quinones MA. Important advances in technology: echocardiography. Methodist Debakey Cardiovasc J. 2014;10(3):146-151.
doi pubmed - Grimard BH, Safford RE, Burns EL. Aortic stenosis: diagnosis and treatment. Am Fam Physician. 2016;93(5):371-378.
pubmed - Baumgartner H, Hung J, Bermejo J, Chambers JB, Edvardsen T, Goldstein S, Lancellotti P, et al. Recommendations on the echocardiographic assessment of aortic valve stenosis: a focused update from the european association of cardiovascular imaging and the American Society of Echocardiography. J Am Soc Echocardiogr. 2017;30(4):372-392.
doi pubmed - Bhatia A. Transesophageal echocardiography evaluation of tricuspid and pulmonic valves. Ann Card Anaesth. 2016;19(Supplement):S21-S25.
doi pubmed - Hoedemakers S, Pugliese NR, Stassen J, Vanoppen A, Claessens J, Gojevic T, Bekhuis Y, et al. mPAP/CO slope and oxygen uptake add prognostic value in aortic stenosis. Circulation. 2024;149(15):1172-1182.
doi pubmed - Bombace S, Meucci MC, Fortuni F, Ilardi F, Manzo R, Canciello G, Esposito G, et al. Beyond aortic stenosis: addressing the challenges of multivalvular disease assessment. Diagnostics (Basel). 2023;13(12):2102.
doi pubmed - Castiglione V, Aimo A, Vergaro G, Saccaro L, Passino C, Emdin M. Biomarkers for the diagnosis and management of heart failure. Heart Fail Rev. 2022;27(2):625-643.
doi pubmed - Katz M, Tarasoutchi F, Pesaro AE, Lopes RD, Spina GS, Vieira ML, Grinberg M. Natriuretic peptides and long-term mortality in patients with severe aortic stenosis. J Heart Valve Dis. 2012;21(3):331-336.
pubmed - Chin CW, Djohan AH, Lang CC. The role of cardiac biochemical markers in aortic stenosis. Biomarkers. 2016;21(4):316-327.
doi pubmed - Wen C, Leiyang Z, Fei D, Yifan Z, Xiao R, Li L, Liang Z, et al. Decreased and inactivated nuclear factor kappa B 1 (p50) in human degenerative calcified aortic valve. Cardiovasc Pathol. 2013;22(1):28-32.
doi pubmed - Ali OA, Chapman M, Nguyen TH, Chirkov YY, Heresztyn T, Mundisugih J, Horowitz JD. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart. 2014;100(10):800-805.
doi pubmed - Jiang Q, Liu SZ, Jiang L, Huang KL, Guo J, Hu SS. Comparison of two radiofrequency ablation devices for atrial fibrillation concomitant with a rheumatic valve procedure. Chin Med J (Engl). 2019;132(12):1414-1419.
doi pubmed - Salem SA, Foerst JR. Valve-in-valve transcatheter aortic valve replacement, with present-day innovations and up-to-date techniques. Interv Cardiol Clin. 2021;10(4):491-504.
doi pubmed - Renker M, Charitos EI, Choi YH, Sossalla S. [Catheter-based and surgical treatment for aortic valve diseases]. Inn Med (Heidelb). 2024;65(5):431-438.
doi pubmed - Elkoumy A, Mylotte D, Elzomor H, McInerney A, Soliman O. Emerging transcatheter heart valve technologies for severe aortic stenosis. Expert Rev Med Devices. 2023;20(12):1065-1077.
doi pubmed - Genereux P, Schwartz A, Oldemeyer JB, Pibarot P, Cohen DJ, Blanke P, Lindman BR, et al. Transcatheter aortic-valve replacement for asymptomatic severe aortic stenosis. N Engl J Med. 2025;392(3):217-227.
doi pubmed - Goody PR, Hosen MR, Christmann D, Niepmann ST, Zietzer A, Adam M, Bonner F, et al. Aortic valve stenosis: from basic mechanisms to novel therapeutic targets. Arterioscler Thromb Vasc Biol. 2020;40(4):885-900.
doi pubmed - Di Fusco SA, Borrelli N, Poli S, Bernelli C, Perone F, Aquilani S, Maggioni AP, et al. [Degenerative aortic valve stenosis: looking for a pharmacological prevention]. G Ital Cardiol (Rome). 2023;24(4):293-304.
doi pubmed - Theriault S, Li Z, Abner E, Luan J, Manikpurage HD, Houessou U, Zamani P, et al. Integrative genomic analyses identify candidate causal genes for calcific aortic valve stenosis involving tissue-specific regulation. Nat Commun. 2024;15(1):2407.
doi pubmed - Albert BJ, Butcher JT. Future prospects in the tissue engineering of heart valves: a focus on the role of stem cells. Expert Opin Biol Ther. 2023;23(6):553-564.
doi pubmed - Spilias N, Martyn T, Denby KJ, Harb SC, Popovic ZB, Kapadia SR. Left ventricular systolic dysfunction in aortic stenosis: pathophysiology, diagnosis, management, and future directions. Struct Heart. 2022;6(5):100089.
doi pubmed - Jiang Q, Yu T, Huang K, Huang X, Zhang Q, Hu S. The impact of medical insurance reimbursement on postoperative inflammation reaction in distinct cardiac surgery from a single center. BMC Health Serv Res. 2022;22(1):494.
doi pubmed - Katsi V, Magkas N, Antonopoulos A, Trantalis G, Toutouzas K, Tousoulis D. Aortic valve: anatomy and structure and the role of vasculature in the degenerative process. Acta Cardiol. 2021;76(4):335-348.
doi pubmed - Chong T, Lan NSR, Courtney W, He A, Strange G, Playford D, Dwivedi G, et al. Medical therapy to prevent or slow progression of aortic stenosis: current evidence and future directions. Cardiol Rev. 2024;32(6):473-482.
doi pubmed - Jiang Q, Huang K, Yin L, Zhang B, Wang Y, Hu S. Multimodality Cardiovascular Imaging for Totally Video-Guided Thorascopic Cardiac Surgery. Rev Cardiovasc Med. 2024;25(5):181.
doi pubmed - Jannasch A, Schnabel C, Galli R, Faak S, Buttner P, Dittfeld C, Tugtekin SM, et al. Optical coherence tomography and multiphoton microscopy offer new options for the quantification of fibrotic aortic valve disease in ApoE(-/-) mice. Sci Rep. 2021;11(1):5834.
doi pubmed - Pappalardo O, Benfari G, Jenkins W, Foley T, Araoz P, Redaelli A, Onorati F, et al. Quantification of anatomical aortic valve area by multi-detector computed tomography: A pilot 3D-morphological modeling of the stenotic aortic valve. Int J Cardiol. 2024;413:132322.
doi pubmed - Nordquist E, LaHaye S, Nagel C, Lincoln J. Corrigendum: postnatal and adult aortic heart valves have distinctive transcriptional profiles associated with valve tissue growth and maintenance respectively. Front Cardiovasc Med. 2019;6:164.
doi pubmed - Martin-Rojas T, Mourino-Alvarez L, Alonso-Orgaz S, Rosello-Lleti E, Calvo E, Lopez-Almodovar LF, Rivera M, et al. iTRAQ proteomic analysis of extracellular matrix remodeling in aortic valve disease. Sci Rep. 2015;5:17290.
doi pubmed - Dutton LC, Dudhia J, Guest DJ, Connolly DJ. CRISPR/Cas9 gene editing in induced pluripotent stem cells to investigate the feline hypertrophic cardiomyopathy causing MYBPC3/R820W mutation. PLoS One. 2024;19(10):e0311761.
doi pubmed - Pavlova SV, Shulgina AE, Zakian SM, Dementyeva EV. Studying pathogenetic contribution of a variant of unknown significance, p.M659I (c.1977G > A) in MYH7, to the development of hypertrophic cardiomyopathy using CRISPR/Cas9-engineered isogenic induced pluripotent stem cells. Int J Mol Sci. 2024;25(16):8695.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.