| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://cr.elmerpub.com |
Review
Volume 16, Number 3, June 2025, pages 169-177

A Review on the Role of DNA Methylation in Aortic Disease Associated With Marfan Syndrome
Wei Ze Zhanga, Chen Ye Wua, Hao Laia, b
aDepartment of Cardiac Surgery, Zhongshan Hospital, Fudan University, Shanghai, China
bCorresponding Author: Hao Lai, Department of Cardiac Surgery, Zhongshan Hospital, Fudan University, Shanghai 200032, China
Manuscript submitted December 23, 2024, accepted March 8, 2025, published online April 22, 2025
Short title: DNA Methylation in Aortic Disease of MFS
doi: https://doi.org/10.14740/cr2033
| Abstract | ▴Top |
Marfan syndrome (MFS) is a genetic disorder primarily affecting the connective tissue, with cardiovascular complications as the leading cause of mortality. While mutations in the FBN1 gene are the primary cause, the severity and progression of the disease can vary significantly among individuals. DNA methylation, a key epigenetic regulatory mechanism, has garnered attention in MFS research, particularly regarding methylation changes in the FBN1 locus and their effects on fibrillin-1 expression. Differential methylation and expression of genes related to inflammation (e.g., interleukin (IL)-10, IL-17) and oxidative stress (e.g., PON2, TP53INP1) have been linked to MFS aortic pathology. These alterations likely contribute to disease progression by influencing inflammatory responses, smooth muscle cell apoptosis, and biomechanical properties of the aorta. The transforming growth factor-beta (TGF-β) signaling pathway plays a central role in MFS pathology, with aberrant methylation of related genes potentially elevating active TGF-β levels and exacerbating aortic lesions. Notably, tissue-specific methylation patterns, especially in smooth muscle cells of the aorta, remain poorly understood. A deeper understanding of DNA methylation’s role in MFS could pave the way for early interventions and epigenetic-targeted therapies.
Keywords: Marfan syndrome; FBN1; DNA methylation; Aortic disease
| Introduction | ▴Top |
Marfan syndrome (MFS) is a heritable connective tissue disorder that affects multiple systems, including the cardiovascular, skeletal, and ocular systems. It is primarily caused by mutations in the FBN1 gene, which encodes fibrillin-1, a crucial component of extracellular matrix microfibrils essential for maintaining tissue structure and elasticity, as well as regulating the bioavailability of transforming growth factor-beta (TGF-β) [1, 2]. Although genetic mutations provide the foundation for disease development, the observed phenotypic variability underscores the involvement of additional regulatory mechanisms, such as epigenetics. DNA methylation is a key epigenetic modification that involves the addition of a methyl group to cytosine bases, particularly within CpG dinucleotides. It influences gene expression, with promoter methylation often associated with gene silencing [3], while methylation within gene bodies and other regulatory regions has more complex effects [4].
Current research on DNA methylation in MFS primarily focuses on investigating its impact on FBN1 expression and related influenced genes or pathways. Existing studies demonstrate significant variations in fibrillin-1 expression levels across different tissues [5], potentially attributable to varying CpG methylation levels; however, previous genome-wide methylation analyses have not revealed altered FBN1 locus methylation levels in aortic tissues from MFS patients, although limitations in control group selection and sample sources, such as peripheral blood leukocytes, constrain the interpretability of these findings [6, 7]. Several studies suggest a correlation between hypomethylation upstream of the FBN1 promoter and elevated gene expression, with the use of demethylation inhibitors shown to increase fibrillin-1 protein expression [8, 9]. Furthermore, other investigations have identified altered methylation levels in genes related to inflammatory responses, oxidative stress, mitochondrial dysfunction, and mechanical stress responses in MFS patients, suggesting a potential role for DNA methylation in MFS pathogenesis through the modulation of multiple pathways [10-12]. Therefore, in-depth investigation of methylation patterns within different regulatory regions of the FBN1 gene, as well as methylation changes in other relevant genes, is crucial for elucidating the pathogenic mechanisms of MFS and developing novel therapeutic strategies.
| Genetic Basis of MFS | ▴Top |
FBN1, fibrillin-1, and TGF-β
The FBN1 gene, located on the long arm of chromosome 15 at the 21.1 band, comprises 65 exons that transcribe into a 10 kb mRNA, which is then translated into the 2,871 amino acid protein fibrillin-1. To date, over 3,000 mutation sites have been identified on this gene, encompassing a wide array of variant types [2, 13]. Approximately 75% of MFS patients have a family history with the same genetic mutation, indicating a strong hereditary component. However, about 25% of cases arise from de novo mutations that occur during gametogenesis or are the result of spontaneous mutations [14].
Fibrillin-1, the expression product of the FBN1 gene, is an extracellular matrix glycoprotein that can polymerize to form microfibril structures, constituting an essential part of the extracellular matrix [15]. The composition and structure of fibrillin-1 are tissue-specific. It can exist independently in tissues such as the periosteum, zonular fibers of the lens, sclera, and cornea; it also allows elastin to deposit on microfibril bundles during development and growth to provide stability and elasticity to tissues that require contraction and expansion, as seen in arteries, lungs, and skin [2, 16]. In the aortic tunica media, abundant elastin-rich microfibril bundles (ERMB) serve as a force-transmitting bridge between smooth muscle cells (SMCs) and the extracellular matrix [2].
Fibrillin-1 not only plays a role in maintaining the structural integrity and mechanical force transduction of the aortic wall but also interacts with numerous extracellular matrix components, among which TGF-β is of paramount importance. TGF-β is secreted by endothelial cells and connects with microfibrils in the form of a complex. These microfibrils serve as a reservoir for TGF-β, regulating its release into the extracellular matrix to act on target cells and activate the TGF-β receptor pathway [17-19]. An anomaly in quantity or structure of fibrillin-1 disrupts its function in biological scaffolding and in sequestration of TGF-β, thereby activating the classical TGF-β signaling pathway [20], which regulates transcription through Smad2-Smad4 complex and induces the production of matrix metalloproteases (MMPs) that degrade the extracellular matrix [21, 22]. Additionally, the aberrant TGF-β causes disarray in the alignment of SMCs, generates ROS inducing vSMC senescence and apoptosis [20, 23], playing a significant role in the development and progression of aortic disease in MFS patients [24-27].
FBN1 mutation classifications
Based on the mutation types, the effect of FBN1 gene mutations can be broadly classified into two categories [28, 29]: 1) dominant negative (DN): mutant FBN1 genes transcribe and translate fibrillin-1 proteins with abnormal structures that interfere with the products from the wild-type allele; 2) haploinsufficiency (HI): the transcribed truncated mRNA from the mutant FBN1 gene is degraded by the nonsense-mediated mRNA decay (NMD) mechanism and is not translated into protein, leading to a reduction of about half in the extracellular matrix fibrillin-1 production. Although no dedicated statistical analysis has been conducted, related studies on gene sequencing in MFS patients suggest that HI accounts for approximately 40% of MFS cases [30, 31]. Generally speaking, the clinical manifestations of protein products caused by FBN1 gene mutations are more severe for HI than DN. Clinical data studies (including a retrospective study involving more than 1,500 MFS patients) have shown that the clinical manifestations caused by HI, such as lens dislocation, skeletal developmental deformities, aortic diameter and expansion rate, and the incidence of aortic dissection represent more severe than DN [32-37]. Experimentally, Aubart et al found that fibrillin-1 level was inversely correlated with the severity of lens dislocation, thoracic deformities, and the Z-score of the aorta [32]. Jondeau et al introduced a WT gene into the FBN1C1039G/- mouse heterozygous background (responsible for increasing the production of normal FBN1), rescuing the vascular phenotype in the MFS mouse model, indicating that fibrillin-1 protein levels can alleviate the clinical manifestations of MFS to a certain extent [38].
Given the intricate relationship between FBN1 gene expression levels and the clinical manifestations of MFS, it is plausible to consider therapeutic strategies that aim to restore normal fibrillin-1 levels. One study has shown that reducing the degradation of aberrant mRNA for increasing the expression levels of the mutated FBN1 gene yields some beneficial effects in HI mutational type; however, this therapeutic method is associated with cytotoxic effects for DN mutational type due to the potential intervention of the aberrant product to the function of fibrillin-1 [29].
Therefore, we propose a hypothesis that a promising approach is to upregulate the expression of the FBN1 gene through DNA methylation, a key epigenetic mechanism, thereby increasing the synthesis of functional fibrillin-1 from normal FBN1 allelic gene against the effects of HI mutational type. Consequently, adjustment of DNA methylation profiles may offer a novel approach to ameliorate the vascular and skeletal abnormalities associated with MFS, ultimately improving patient outcomes.
| DNA Methylation | ▴Top |
CpG islands (CGIs) and DNA methylation
CpG dinucleotides, which consist of cytosine and guanine linked by a phosphodiester bond, make up less than 1% of the human genome. However, they tend to cluster in regions called CGIs, which are typically defined as DNA sequences of at least 200 base pairs with a GC content exceeding 50% [39]. Approximately 29,000 CGIs are identified in the human genome, predominantly located near transcription start sites or within promoter regions associated with the first exon. In mammalian genomes, around 70% of CGIs are constitutively methylated, whereas cytosines in CGIs located within promoters typically remain hypomethylated [40, 41].
DNA methylation is catalyzed by DNA methyltransferases (DNMTs), which transfer a methyl group from S-adenosylmethionine (SAM) to the fifth carbon of the cytosine ring, forming 5-methylcytosine (5mC) [42]. The DNMT family in mammals comprises enzymatically active DNMT1, DNMT3A, and DNMT3B, along with DNMT2 and DNMT3L. DNMT1 recognizes hemimethylated CpG dinucleotides (where methylation occurs on only one DNA strand) and methylates the complementary strand, ensuring the maintenance of methylation patterns during DNA replication. This process is essential for preserving methylation stability in progeny cells. DNMT3A and DNMT3B, on the other hand, are responsible for de novo methylation and establish genome-wide methylation patterns during the preimplantation blastocyst stage. Subsequently, DNMT1 maintains these patterns during cell division, ensuring their stability and fidelity [43]. During development, cell-specific gene expression is regulated by the coordinated activity of ten-eleven translocation (TET) enzymes, which play a key role in active DNA demethylation, along with DNMT3A and DNMT3B [44]. Additionally, DNMT2 primarily catalyzes tRNA methylation [45], while DNMT3L, despite lacking catalytic activity, enhances the function of DNMT3A and DNMT3B [46].
DNA demethylation occurs via two primary mechanisms. Passive demethylation arises when DNMT1 fails to maintain methylation during replication, leading to progressive dilution of methylation marks over successive cell divisions [47]. Active demethylation is achieved through the sequential oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) by TET enzymes [48]. These intermediates are subsequently removed through base excision repair pathways [49].
DNA methylation regulation and inhibitors
Based on their proximity to CGIs, methylation sites are categorized into CpG shores (within 2 kb of a CGI), CpG shelves (2 - 4 kb from a CGI), and open seas (the remaining genomic regions). Methylation levels in these regions vary and can significantly impact gene expression [50-52]. For instance, studies in the field of MFS have reported associations between CpG shore methylation and transcriptional repression [8]. Research on DNA methylation has traditionally focused on promoter methylation but is now expanding to include gene body, enhancer, and insulator methylation [53, 54].
Generally, hypermethylation of promoter regions represses gene expression, whereas hypomethylation facilitates it. Mechanistically, this can occur through the recruitment of methyl-CpG-binding domain (MBD) proteins, which block transcription factors from binding to methylated DNA, or through methylation-dependent recruitment of repressive chromatin remodelers, leading to heterochromatin formation and transcriptional silencing [55]. However, an increasing number of studies indicate that promoter hypermethylation may also correlate with elevated expression of certain genes, depending on factors such as the specific methylated region or the number of methylated CpG sites. For instance, studies have reported the number of core methylation sites in promoters of the same gene as critical role for regulating gene expression range from 1 to 52 [56-58]. While the density of CpGs does not always directly correlate with transcriptional regulation, defining the range of methylation sites within promoter regions remains a critical consideration.
Gene body methylation (GBM) is a widely conserved epigenetic modification. In the human genome, while CpG sites predominantly cluster in promoter regions, 40-50% are located within gene bodies, where methylation is more prevalent [59, 60]. Typically, promoter hypermethylation suppresses transcription, whereas gene body hypermethylation often correlates with active transcription [61, 62]. However, a meta-analysis revealed that the relationship between GBM levels and gene expression is not linear but spindle-shaped, with both low and high levels of GBM reducing gene expression, while moderate methylation enhances it. The underlying mechanisms may include not only the suppression of transcriptional isoforms driven by intragenic alternative promoters and the regulation of splicing but also the spatial facilitation of RNA polymerase binding and its interaction with chromatin accessibility [59, 61, 63]. Exons tend to have higher methylation levels than introns, with more extensive methylation in housekeeping genes than in regulatory genes [64, 65], and tissue-specific methylation patterns are more commonly observed in intragenic CGIs [66].
DNA methylation inhibitors are broadly categorized into nucleoside analogs and non-nucleoside analogs. Nucleoside analogs include 5-azacytidine (5-aza), 5-aza-2’-deoxycytidine (decitabine, DAC), zebularine, and 5-fluoro-2’-deoxycytidine (FdCyd). Non-nucleoside analogs include procaine, procainamide, nanaomycin A, hydralazine, valproic acid, and natural compounds such as epigallocatechin-3-gallate (EGCG), genistein, and curcumin [41]. Among these, 5-aza and DAC are the most commonly used and have been approved by the European Medicines Agency and the United States Food and Drug Administration (FDA) for treating acute myeloid leukemia (AML), myelodysplastic syndromes, and chronic myelomonocytic leukemia [41, 67]. These agents are incorporated into DNA during the S phase of the cell cycle as active triphosphate nucleotides, forming covalent bonds with DNMTs and inhibiting their activity, thereby inducing DNA demethylation [68]. 5-aza reduces DNA methylation levels, influencing cellular phenotypes by increasing the expression of genes associated with the differentiated SMC phenotype and suppressing inflammatory factors [68]. It also inhibits fibroblast-to-myofibroblast differentiation in the presence of TGF-β, thereby reducing tissue fibrosis [69]. DAC exhibits approximately 90% higher demethylation efficiency than 5-aza [70]. However, both agents are non-specific DNMT inhibitors and are susceptible to degradation by cytidine deaminase (CDA), which is abundant in the gastrointestinal tract, leading to reduced bioavailability. Parenteral administration can mitigate this issue but exacerbates side effects. Combining DAC with cedazuridine, a CDA inhibitor, has enabled oral administration and received FDA approval for clinical use [71]. Zebularine and FdCyd, as optimized derivatives of 5-aza, exhibit reduced CDA-mediated degradation, lower toxicity, and improved tissue specificity [72, 73].
| Exploring DNA Methylation in MFS | ▴Top |
Of greatest interest and concern for the study of DNA methylation alterations within the field of MFS is whether the FBN1 locus is specifically methylated and altered in MFS relative to degenerative aneurysms and whether this results in changes in fibrillin-1 protein expression. It was shown that fibrillin-1 expression levels vary significantly in different tissues and organs, and a 4.4-fold interindividual difference in FBN1 mRNA was observed in skin fibroblasts from MFS patients, which may be attributed to the differential methylation levels of CpG [9, 32]. Dong analyzed the whole-genome methylation levels of aortic tissues of MFS patients and controls and the results did not reveal altered methylation levels of the FBN1 locus. However, the control group selected for this study used age and sex as matching variables rather than patients with aortic aneurysms, so this is not sufficient evidence in response to the results of MFS-specific methylation alterations [6]. Malecki also did not find methylation changes in the upstream and internal regions of the FBN1 gene by examining peripheral blood leukocyte methylation levels. It should be noted that the methylation levels of peripheral blood leukocytes used in their study did not accurately match the specific methylation changes in diseased aortic tissues [7]. Arai et al studied FBN1 single-gene methylation levels in porcine embryonic cells and fibroblasts and found that the proportion of hypomethylated alleles (the ratio of unmethylated sites to total measured sites in CpG banks) upstream of the FBN1 promoter was significantly and positively correlated with gene expression levels. Treatment with the demethylation inhibitor 5-aza reduced the methylation level of the FBN1 promoter region and increased fibrillin-1 protein expression, suggesting that promoter methylation inhibits FBN1 expression. Examination of the promoter region of the FBN1 gene in human cell lines on MRC5, AM936EP, Edom22, PAE551, and UtE1104 revealed heterogeneous levels of CGI shore methylation, but these cell lines were not derived from aortic tissue [8, 9]. The level of fibrillin-1 protein expression is primarily influenced by the methylation status of the wild-type FBN1 allele. In contrast, the methylation level of the transcription product of the haploinsufficient allele has little effect on fibrillin-1 expression, as it is eliminated by NMD during splicing or translation. However, high expression of the dominant-negative allele may exacerbate the symptoms of MFS [8, 29].
In addition, DNMTs and TETs-related enzyme families may play an important role in the methylation level of the FBN1 locus during embryonic developmental stages. It has now been shown that demethylation and remethylation of the FBN1 locus occurs from porcine gametes to blastocysts to mulberry embryos [9]. Passaging culture experiments with FBN1 knockout fibroblasts have shown that the methylation level of the FBN1 promoter increases with the number of passages, leading to a decrease in the mRNA/preRNA ratio [8]. It is worth mentioning that SMCs from diseased aortas derived from MFS have not been studied by DNA methylation methods alone. Studies have shown that aortic SMCs with the ability of secreting fibrillin-1, and mice with specific FBN1 gene knockout in SMC can actively induce aortic aneurysm in mice [74]. Our study and related researches demonstrated that abnormal fibrillin-1 expression leads to abnormal TGF-β pathway and induces SMC apoptosis [20, 75], and this vicious cycle probably contributes to the occurrence of aortic lesions in MFS. In addition, one hypothesis is proposed that the different levels of methylation formed on the WT-type FBN1 allele during early developmental stages can cause differences in fibrillin-1 output, thus contributing to the phenomenon of heterogeneity in the timing of aortic lesion onset in patients with the same causative gene within the MFS family [8].
The researches about whether methylation sites located in different DNA regulatory sequences affect FBN1 gene expression in different ways are limited. Arai et al focused on the CGI shore in the promoter region [9], while Dong and Malecki found that most of the CpG methylation differences detected at the global genome level were distributed within the gene body, with a few located in the promoter region and the 3'UTR [6, 7]. Holden et al analyzed the methylation distribution of the 65 exons of the FBN1 gene, and found that the highest proportions of CpG dinucleotides were found in exon 1 (9%), exon 44 (6.5%), exon 24 (5.3%), and exon 27 (4.8%), all of which significantly exceed the CpG proportion of approximately 1% of the human genome [76]. Some evidence suggests that the strongest correlation between methylation and transcription is not located at the gene promoter but up to 8 kilobases (kb) downstream of the promoter, specifically within the gene (0.3 - 8 kb) [77]. In our previous study, we collected aortic tissues from patients with aortic dissection and healthy controls and performed Illumina DNA methylation profiling, observing significant non-CpG site methylation in normal aorta as well as a global loss of DNA methylation in the non-CGI region of patients [78]. All of this evidence suggests that the methylation sites regulating FBN1 gene expression are not only located in the promoter, but are more likely to be located within other DNA regulatory sequences yet to be discovered.
In addition to the possible methylation alterations and therapeutic targets of the FBN1 locus, the expression levels and potential alterations in methylation levels of other minor loci that may affect the MFS phenotype deserve further investigation. Methylation assays for both peripheral blood leukocytes and aortic tissues from patients with MFS demonstrated that genes corresponding to methylation-altered loci were predominantly enriched in inflammatory responses, oxidative stress, mitochondrial dysfunction, and mechanical stress responses. Among them, the expression of the inflammatory cytokines interleukins (IL) IL-10, IL-17, IL-36g, and IL-38 was upregulated, a finding validated by immunofluorescence of MFS aortic tissue [10-12]. The corresponding CpG site with the highest altered methylation levels was located in the 3'UTR region of IL-17RA and correlated with the severity of the clinical phenotype [12, 79]. In addition, increased promoter methylation of PON2, CYP1A1, and TP53INP1 involved in oxidative stress and decreased methylation of FLJ44606 correlated with increased aortic diameter, worsening of left ventricular ejection fraction (LVEF), and increased left ventricular end-diastolic diameter (LVEDD) [10]. Van Andel et al found that aorta root diameter of patients with MFS was negatively correlated with methylation levels at the cg00702593 locus, corresponding to the SAMD4A gene, whose expression product inhibits angiogenesis and is involved in myopathy. Methylation levels of the MEF2D and LOC100506 genes were positively and negatively correlated, respectively, with aortic events (all-cause mortality, aortic coarctation/rupture, or surgery) [80]. DNA methylation may contribute to aortic events by affecting the expression of numerous genes leading to aberrant pathway changes that ultimately affect the phenotype of MFS. Liddy et al found that most proteins (65%) in the TGF-β pathway exhibited significantly reduced CpG site methylation, primarily in the promoter region, when analyzing peripheral blood leukocytes from MFS patients. This suggests that aberrant methylation affects the expression of genes related to the TGF-β pathway in MFS [81]. Additionally, high levels of active TGF-β have been detected in the extracellular medium of MFS SMCs, which may be attributed to epigenetic modifications [82]. Dong used aortic tissues from MFS patients to identify six pathways associated with differentially methylated sites through KEGG analysis. These pathways include HIF-1, JAK-STAT, cGMP-PKG, sphingolipid, Rap1, and PI3K/AKT [6]. Among them, the PI3K/AKT signaling pathway has multiple cross-linking effects, including the ability to induce SMC phenotypic transformation, improve vasodilatory function, and prevent aortic aneurysm formation [83-85]. Notably, during aneurysm formation, the loss of elastin and the deposition and increased cross-linking of collagen translate into biomechanical changes, including reduced distensibility and increased stiffness of the aorta [86]. Studies have shown that MFS patients exhibit a compensatory elevation of lysyl oxidases (LOX), an extracellular enzyme that facilitates the covalent cross-linking of collagen and elastin, promoting extracellular matrix maturation. This is evidenced by increased aortic strength and a reduced risk of rupture after aortic dilatation in both MFS patients and a mouse model. When an LOX inhibitor was administered in the mouse model, aortic stiffness and strength were reduced, leading to an increased risk of rupture after aortic dilatation [87, 88]. In addition, the histopathological features of MFS aneurysm tissues include elastic lamellae fragmentation and disintegration, excessive apoptosis of SMCs, and increased expression of MMP-2 and MMP-9 [89], all of which have been shown to increase the risk of aortic disease. The potential regulation of DNA methylation behind the phenomenon of elevated Lox and MMPs in MFS has not yet been investigated.
According to the key findings discussed in this review, Figure 1 outlines potential future directions for research in this field, which could focus on conducting detailed analyses of methylation sites and regions within regulatory elements or the gene body of FBN1 gene with HI mutational type; investigating whether DNMTs, TETs, and other methylation-catalytic enzymes exhibit alterations in methylation patterns or expression levels during early developmental stages in individuals with MFS; and exploring additional potential methylation changes in genes that may influence the clinical symptoms observed in MFS patients.
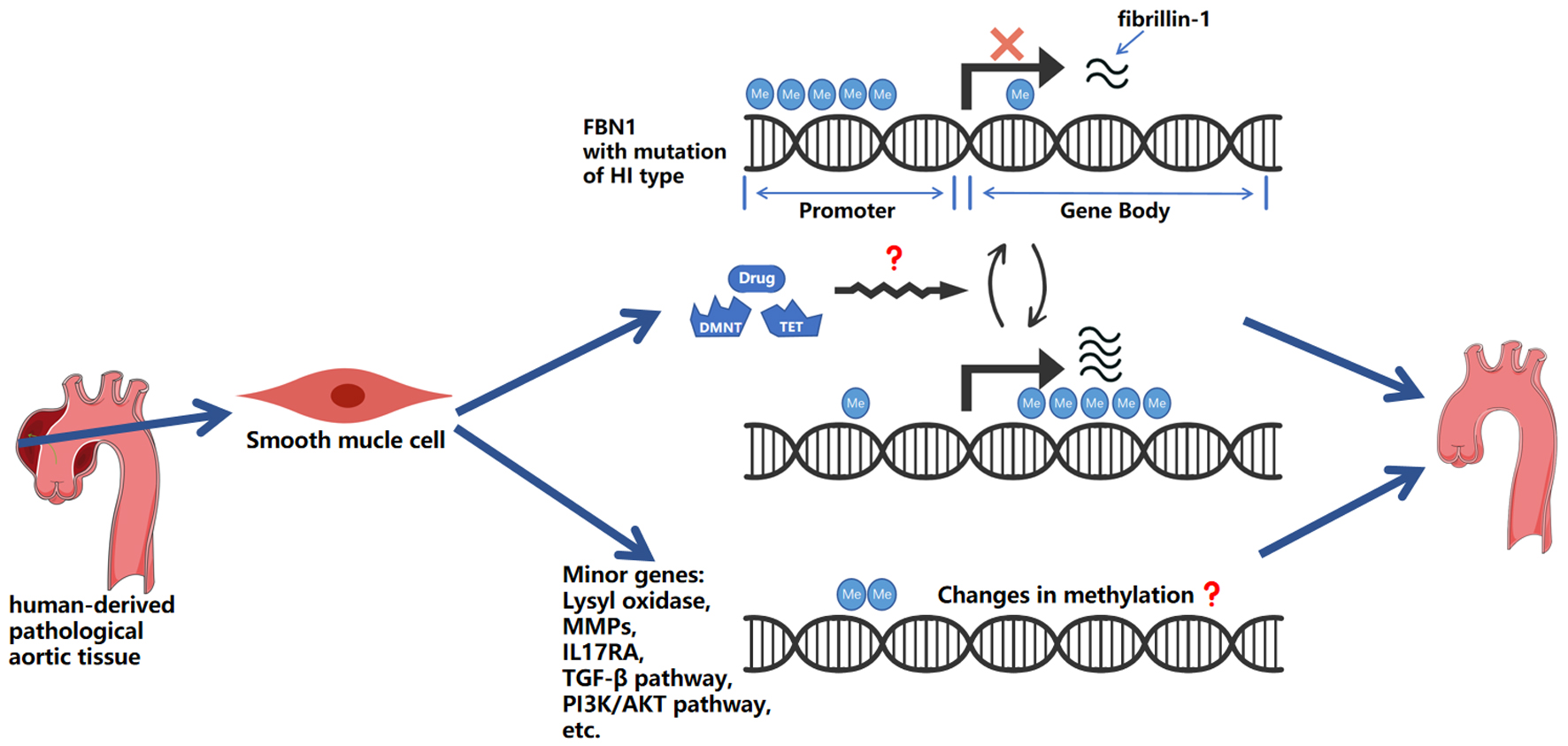 Click for large image | Figure 1. Schematic representation of hypothesized mechanisms linking DNA methylation to aortic aneurysm progression. This figure highlights: 1) the potential impact of gene body hypermethylation on gene expression, and the complex relationship between promoter and gene body methylation in regulating FBN1 with HI mutational type; 2) the potential of DNMT and TET enzymes, and other methylation-modifying substances, as research tools and therapeutic agents; and 3) the involvement of lysyl oxidases, MMPs, IL-17RA, and genes within the TGF-β and PI3K/AKT pathways, including other potentially methylation-regulated genes. Me: methylation; HI: haploinsufficiency; TGF-β: transforming growth factor-beta; MMPs: matrix metalloproteases; DNMT: DNA methyltransferases; TET: ten-eleven translocation. |
| Conclusion and Perspective | ▴Top |
Heterogeneity in the timing of onset and severity of clinical symptoms among MFS patients with the same causative gene in the family has long been of great interest. The current study shows that the type of gene variant is not clearly associated with the clinical symptoms of MFS, and that the clinical symptoms of MFS correlate with the number of structurally intact FBN1 gene products. DNA methylation, as an important branch of epigenetics, has been investigated to explore its effect on MFS. Currently, correlation studies of methylation alterations in the promoter region of the FBN1 gene, secondary genes and related genes of molecular pathways have initially demonstrated the great potential of DNA methylation as a therapeutic target for MFS. By addressing the challenges, coupled with the development of methylation-targeted drugs, the future prospect of utilizing drug-based approaches as a potential treatment for MFS is promising, though further research is needed to validate its efficacy.
Acknowledgments
None to declare.
Financial Disclosure
No funding was provided for this manuscript.
Conflict of Interest
The authors declare no competing interest.
Author Contributions
Hao Lai provided writing direction, critical feedback, and revisions. Wei Ze Zhang conducted the literature search, reviewed the articles, and drafted the manuscript. Chen Ye Wu contributed ideas and suggestions that enhanced the scope of the review. All authors approved the final manuscript.
Data Availability
This review is based on publicly available data and literature from Google Scholar and Pubmed, which are cited in the references. The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
AML: acute myeloid leukemia; CDA: cytidine deaminase; CGIs: CpG islands; DAC: 5-aza-2’-deoxycytidine; DN: dominant negative; DNMTs: DNA methyltransferases; EGCG: epigallocatechin-3-gallate; ERMB: elastin-rich microfibril bundles; 5-aza: 5-azacytidine; 5caC: 5-carboxylcytosine; 5fC: 5-formylcytosine; 5hmC: 5-hydroxymethylcytosine; 5mC: 5-methylcytosine; FdCyd: 5-fluoro-2’-deoxycytidine; GBM: gene body methylation; HI: haploinsufficiency; IL: interleukin; LVEDD: left ventricular end-diastolic diameter; LVEF: left ventricular ejection fraction; MBD: methyl-CpG-binding domain; MFS: Marfan syndrome; MMPs: matrix metalloproteases; NMD: nonsense-mediated mRNA decay; SAM: S-adenosylmethionine; SMCs: smooth muscle cells; TET: ten-eleven translocation; TGF-β: transforming growth factor-beta
| References | ▴Top |
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-485.
doi pubmed - Milewicz DM, Braverman AC, De Backer J, Morris SA, Boileau C, Maumenee IH, Jondeau G, et al. Marfan syndrome. Nat Rev Dis Primers. 2021;7(1):64.
doi pubmed - Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457-466.
doi pubmed - Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484-492.
doi pubmed - Davis MR, Arner E, Duffy CR, De Sousa PA, Dahlman I, Arner P, Summers KM. Expression of FBN1 during adipogenesis: Relevance to the lipodystrophy phenotype in Marfan syndrome and related conditions. Mol Genet Metab. 2016;119(1-2):174-185.
doi pubmed - Dong CX. Molecular mechanisms of thoracic aortic aneurysm in Marfan Syndrome. 2023.
- Malecki C. Modulators of phenotypic variation associated with genetically triggered thoracic aortic aneurysms. 2021.
- Arai Y, Umeyama K, Okazaki N, Nakano K, Nishino K, Nagashima H, Ohgane J. DNA methylation ambiguity in the Fibrillin-1 (FBN1) CpG island shore possibly involved in Marfan syndrome. Sci Rep. 2020;10(1):5287.
doi pubmed - Arai Y, Umeyama K, Takeuchi K, Okazaki N, Hichiwa N, Yashima S, Nakano K, et al. Establishment of DNA methylation patterns of the Fibrillin1 (FBN1) gene in porcine embryos and tissues. J Reprod Dev. 2017;63(2):157-165.
doi pubmed - Malecki C, et al. Changes in DNA methylation indicate a role of oxidative stress in Marfan syndrome. Arteriosclerosis, Thrombosis, and Vascular Biology. 2019;39(Suppl_1):A644-A644.
- Malecki C, et al. Differential DNA methylation in Marfan syndrome is associated with inflammation and cardiac contraction. Circulation. 2019;140(Suppl_1):A14231-A14231.
- Malecki C, et al. DNA methylation of interleukin-17 receptor correlates with disease severity in Marfan syndrome. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(Suppl_1):A405-A405.
- De Backer J, Campens L, Muino Mosquera L. Looking for the missing links: challenges in the search for genotype-phenotype correlation in Marfan syndrome. Circ Genom Precis Med. 2018;11(6):e002185.
doi pubmed - Canadas V, Vilacosta I, Bruna I, Fuster V. Marfan syndrome. Part 1: pathophysiology and diagnosis. Nat Rev Cardiol. 2010;7(5):256-265.
doi pubmed - Asano K, Cantalupo A, Sedes L, Ramirez F. The multiple functions of fibrillin-1 microfibrils in organismal physiology. Int J Mol Sci. 2022;23(3):1892.
doi pubmed - Sakai LY, Keene DR, Renard M, De Backer J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene. 2016;591(1):279-291.
doi pubmed - Lockhart-Cairns MP, Cain SA, Dajani R, Steer R, Thomson J, Alanazi YF, Kielty CM, et al. Latent TGFbeta complexes are transglutaminase cross-linked to fibrillin to facilitate TGFbeta activation. Matrix Biol. 2022;107:24-39.
doi pubmed - De Backer J, et al. Poster No. 133 Local TGF-beta sequestration by fibrillin-1 regulates vascular wall homeostasis in the thoracic aorta. Cardiovascular Research. 2022;118(Supplement_2):cvac157.111.
- Frangogiannis NG. Transforming growth factor-beta in myocardial disease. Nat Rev Cardiol. 2022;19(7):435-455.
doi pubmed - Rombouts KB, van Merrienboer TAR, Ket JCF, Bogunovic N, van der Velden J, Yeung KK. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest. 2022;52(4):e13697.
doi pubmed - Gomez D, Coyet A, Ollivier V, Jeunemaitre X, Jondeau G, Michel JB, Vranckx R. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc Res. 2011;89(2):446-456.
doi pubmed - Gensicke NM, Cavanaugh NB, Andersen ND, Huang T, Qian L, Dyle MC, Turek JW. Accelerated Marfan syndrome model recapitulates established signaling pathways. J Thorac Cardiovasc Surg. 2020;159(5):1719-1726.
doi pubmed - Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. 2017;49(1):97-109.
doi pubmed - Nataatmadja M, West M, West J, Summers K, Walker P, Nagata M, Watanabe T. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation. 2003;108(Suppl 1):II329-334.
doi pubmed - Emrich FC, Okamura H, Dalal AR, Penov K, Merk DR, Raaz U, Hennigs JK, et al. Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(1):146-154.
doi pubmed - Asano K, Cantalupo A, Sedes L, Ramirez F. Pathophysiology and therapeutics of thoracic aortic aneurysm in Marfan syndrome. Biomolecules. 2022;12(1):128.
doi pubmed - Park JH, Kim MS, Ham S, Park ES, Kim KL, Suh W. Transforming growth factor beta receptor type I inhibitor, Galunisertib, has no beneficial effects on aneurysmal pathological changes in Marfan mice. Biomol Ther (Seoul). 2020;28(1):98-103.
doi pubmed - Chen ZX, Jia WN, Jiang YX. Genotype-phenotype correlations of marfan syndrome and related fibrillinopathies: Phenomenon and molecular relevance. Front Genet. 2022;13:943083.
doi pubmed - Balic Z, Hubmacher D. Promoting translational readthrough to augment fibrillin-1 (FBN1) deposition in Marfan syndrome fibroblasts: A proof-of-concept study. bioRxiv. 2022.
- Takeda N, Inuzuka R, Maemura S, Morita H, Nawata K, Fujita D, Taniguchi Y, et al. Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circ Genom Precis Med. 2018;11(6):e002058.
doi pubmed - Hernandiz A, Zuniga A, Valera F, Domingo D, Ontoria-Oviedo I, Mari JF, Roman JA, et al. Genotype FBN1/phenotype relationship in a cohort of patients with Marfan syndrome. Clin Genet. 2021;99(2):269-280.
doi pubmed - Aubart M, Gross MS, Hanna N, Zabot MT, Sznajder M, Detaint D, Gouya L, et al. The clinical presentation of Marfan syndrome is modulated by expression of wild-type FBN1 allele. Hum Mol Genet. 2015;24(10):2764-2770.
doi pubmed - Franken R, Groenink M, de Waard V, Feenstra HM, Scholte AJ, van den Berg MP, Pals G, et al. Genotype impacts survival in Marfan syndrome. Eur Heart J. 2016;37(43):3285-3290.
doi pubmed - Franken R, Teixido-Tura G, Brion M, Forteza A, Rodriguez-Palomares J, Gutierrez L, Garcia Dorado D, et al. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart. 2017;103(22):1795-1799.
doi pubmed - Arnaud P, Milleron O, Hanna N, Ropers J, Ould Ouali N, Affoune A, Langeois M, et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med. 2021;23(7):1296-1304.
doi pubmed - Buki G, Szalai R, Pinter A, Hadzsiev K, Melegh B, Rauch T, Bene J. Correlation between large FBN1 deletions and severe cardiovascular phenotype in Marfan syndrome: Analysis of two novel cases and analytical review of the literature. Mol Genet Genomic Med. 2023;11(7):e2166.
doi pubmed - Polonis K, et al. Haploinsufficient FBN1 variants are associated with a higher risk of aortic complications in pregnant women with Marfan syndrome.
- Jondeau G, Michel JB, Boileau C. The translational science of Marfan syndrome. Heart. 2011;97(15):1206-1214.
doi pubmed - Mattei AL, Bailly N, Meissner A. DNA methylation: a historical perspective. Trends Genet. 2022;38(7):676-707.
doi pubmed - Villicana S, Bell JT. Genetic impacts on DNA methylation: research findings and future perspectives. Genome Biol. 2021;22(1):127.
doi pubmed - Nunes SP, Henrique R, Jeronimo C, Paramio JM. DNA methylation as a therapeutic target for bladder cancer. Cells. 2020;9(8):1850.
doi pubmed - Borchiellini M, Ummarino S, Di Ruscio A. The bright and dark side of DNA methylation: a matter of balance. Cells. 2019;8(10):1243.
doi pubmed - Yagi M, Kabata M, Tanaka A, Ukai T, Ohta S, Nakabayashi K, Shimizu M, et al. Identification of distinct loci for de novo DNA methylation by DNMT3A and DNMT3B during mammalian development. Nat Commun. 2020;11(1):3199.
doi pubmed - Li Z, Dai H, Martos SN, Xu B, Gao Y, Li T, Zhu G, et al. Distinct roles of DNMT1-dependent and DNMT1-independent methylation patterns in the genome of mouse embryonic stem cells. Genome Biol. 2015;16(1):115.
doi pubmed - Jeltsch A, Ehrenhofer-Murray A, Jurkowski TP, Lyko F, Reuter G, Ankri S, Nellen W, et al. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017;14(9):1108-1123.
doi pubmed - Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, Matarese F, et al. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell. 2013;155(1):121-134.
doi pubmed - Cardoso MC, Leonhardt H. DNA methyltransferase is actively retained in the cytoplasm during early development. J Cell Biol. 1999;147(1):25-32.
doi pubmed - Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472-479.
doi pubmed - Duymich CE, Charlet J, Yang X, Jones PA, Liang G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun. 2016;7:11453.
doi pubmed - Ollikainen M, Ismail K, Gervin K, Kyllonen A, Hakkarainen A, Lundbom J, Jarvinen EA, et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin Epigenetics. 2015;7(1):39.
doi pubmed - Visone R, Bacalini MG, Di Franco S, Ferracin M, Colorito ML, Pagotto S, Laprovitera N, et al. DNA methylation of shelf, shore and open sea CpG positions distinguish high microsatellite instability from low or stable microsatellite status colon cancer stem cells. Epigenomics. 2019;11(6):587-604.
doi pubmed - Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178-186.
doi pubmed - Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, Consortium H, et al. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet. 2018;50(3):424-431.
doi pubmed - Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110-114.
doi pubmed - Smith J, Sen S, Weeks RJ, Eccles MR, Chatterjee A. Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer. 2020;6(5):392-406.
doi pubmed - Lindsey JC, Schwalbe EC, Potluri S, Bailey S, Williamson D, Clifford SC. TERT promoter mutation and aberrant hypermethylation are associated with elevated expression in medulloblastoma and characterise the majority of non-infant SHH subgroup tumours. Acta Neuropathol. 2014;127(2):307-309.
doi pubmed - Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013;14(6):534-542.
doi pubmed - Lee DD, Leao R, Komosa M, Gallo M, Zhang CH, Lipman T, Remke M, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. 2019;129(1):223-229.
doi pubmed - Wang Q, Xiong F, Wu G, Liu W, Chen J, Wang B, Chen Y. Gene body methylation in cancer: molecular mechanisms and clinical applications. Clin Epigenetics. 2022;14(1):154.
doi pubmed - Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, Koltsina M, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15(4):r54.
doi pubmed - Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3(4):462-474.
doi pubmed - Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317-330.
doi pubmed - Bacolod MD, Barany F. MGMT epigenetics: the influence of gene body methylation and other insights derived from integrated Methylomic, transcriptomic, and chromatin analyses in various cancer types. Curr Cancer Drug Targets. 2021;21(4):360-374.
doi pubmed - Dixon G, Liao Y, Bay LK, Matz MV. Role of gene body methylation in acclimatization and adaptation in a basal metazoan. Proc Natl Acad Sci U S A. 2018;115(52):13342-13346.
doi pubmed - Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590-607.
doi pubmed - Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253-257.
doi pubmed - Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630-641.
doi pubmed - Strand KA, Lu S, Mutryn MF, Li L, Zhou Q, Enyart BT, Jolly AJ, et al. High Throughput screen identifies the DNMT1 (DNA Methyltransferase-1) inhibitor, 5-azacytidine, as a potent inducer of PTEN (Phosphatase and Tensin Homolog): central role for PTEN in 5-azacytidine protection against pathological vascular remodeling. Arterioscler Thromb Vasc Biol. 2020;40(8):1854-1869.
doi pubmed - Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, Phelan D, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet. 2014;23(8):2176-2188.
doi pubmed - Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85-93.
doi pubmed - Patel AA, Cahill K, Saygin C, Odenike O. Cedazuridine/decitabine: from preclinical to clinical development in myeloid malignancies. Blood Adv. 2021;5(8):2264-2271.
doi pubmed - Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, Marquez VE, et al. Preferential response of cancer cells to zebularine. Cancer Cell. 2004;6(2):151-158.
doi pubmed - Beumer JH, Eiseman JL, Parise RA, Joseph E, Holleran JL, Covey JM, Egorin MJ. Pharmacokinetics, metabolism, and oral bioavailability of the DNA methyltransferase inhibitor 5-fluoro-2'-deoxycytidine in mice. Clin Cancer Res. 2006;12(24):7483-7491.
doi pubmed - Li B, et al. Smooth muscle cell-specific deficiency of fibrillin-1 induces aortic aneurysms but does not disrupt elastic fiber formation in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2024;44(Suppl_1):A2013-A2013.
- Hu C, Tan H, Lin Q, Abudupataer M, Zhao Y, Li J, Gu J, et al. SPECT/CT imaging of apoptosis in aortic aneurysm with radiolabeled duramycin. Apoptosis. 2019;24(9-10):745-755.
doi pubmed - Holden T, et al. Marfan syndrome exon CpG percentage and fractal dimension. In: 2008 2nd International Conference on Bioinformatics and Biomedical Engineering. 2008. IEEE.
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015;523(7559):212-216.
doi pubmed - Pan S, Lai H, Shen Y, Breeze C, Beck S, Hong T, Wang C, et al. DNA methylome analysis reveals distinct epigenetic patterns of ascending aortic dissection and bicuspid aortic valve. Cardiovasc Res. 2017;113(6):692-704.
doi pubmed - Dong CX, Malecki C, Robertson E, Hambly B, Jeremy R. Molecular mechanisms in genetic aortopathy-signaling pathways and potential interventions. Int J Mol Sci. 2023;24(2):1795.
doi pubmed - van Andel MM, Groenink M, van den Berg MP, Timmermans J, Scholte A, Mulder BJM, Zwinderman AH, et al. Genome-wide methylation patterns in Marfan syndrome. Clin Epigenetics. 2021;13(1):217.
doi pubmed - Liddy KA, et al. The pathogenesis of Marfan syndrome involves epigenetic regulation of signalling. Circulation. 2018;138(Suppl_1):A14489-A14489.
- Crosas-Molist E, Meirelles T, Lopez-Luque J, Serra-Peinado C, Selva J, Caja L, Gorbenko Del Blanco D, et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(4):960-972.
doi pubmed - Zhu SB, Zhu J, Zhou ZZ, Xi EP, Wang RP, Zhang Y. TGF-beta1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am J Transl Res. 2015;7(12):2764-2774.
pubmed - Ni XQ, Zhang YR, Jia LX, Lu WW, Zhu Q, Ren JL, Chen Y, et al. Inhibition of Notch1-mediated inflammation by intermedin protects against abdominal aortic aneurysm via PI3K/Akt signaling pathway. Aging (Albany NY). 2021;13(4):5164-5184.
doi pubmed - Lin XP, Cui HJ, Yang AL, Luo JK, Tang T. Astragaloside IV improves vasodilatation function by regulating the PI3K/Akt/eNOS signaling pathway in rat aorta endothelial cells. J Vasc Res. 2018;55(3):169-176.
doi pubmed - Creamer TJ, Bramel EE, MacFarlane EG. Insights on the pathogenesis of aneurysm through the study of hereditary aortopathies. Genes (Basel). 2021;12(2):183.
doi pubmed - Busnadiego O, Gorbenko Del Blanco D, Gonzalez-Santamaria J, Habashi JP, Calderon JF, Sandoval P, Bedja D, et al. Elevated expression levels of lysyl oxidases protect against aortic aneurysm progression in Marfan syndrome. J Mol Cell Cardiol. 2015;85:48-57.
doi pubmed - Weiss D, Rego BV, Cavinato C, Li DS, Kawamura Y, Emuna N, Humphrey JD. Effects of age, sex, and extracellular matrix integrity on aortic dilatation and rupture in a mouse model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2023;43(9):e358-e372.
doi pubmed - Dale M, Fitzgerald MP, Liu Z, Meisinger T, Karpisek A, Purcell LN, Carson JS, et al. Premature aortic smooth muscle cell differentiation contributes to matrix dysregulation in Marfan Syndrome. PLoS One. 2017;12(10):e0186603.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.